

 English
English文献解读|Nat Genet(29.0):KDM4C 抑制通过促进组织蛋白酶 L 介导的组蛋白 H3 裂解来阻止基底乳腺癌中的肿瘤生长
✦ +
+
论文ID
原名:KDM4C inhibition blocks tumor growth in basal breast cancer by promoting cathepsin L-mediated histone H3 cleavage
译名:KDM4C 抑制通过促进组织蛋白酶 L 介导的组蛋白 H3 裂解来阻止基底乳腺癌中的肿瘤生长
期刊:Nature Genetics
影响因子:29.0
发表时间:2025.06.02
DOI号:10.1038/s41588-025-02197-z
背 景
乳腺癌是一种异质性疾病,可分为管腔型、人表皮生长因子受体 2 阳性 (HER2 +) 和基底分子亚型。临床分类基于雌激素 (ER)、孕激素 (PR) 和 HER2 受体的表达,从而区分 ER+、HER2+和三阴性(ER–PR–HER2−) 乳腺癌。三阴性乳腺癌 (TNBC) 通常与治疗耐药性和远处转移的高风险相关,导致患者生存期比其他亚型更短。TNBC 也具有高度异质性,可进一步分为管腔型、基底型和间质型,具有不同的突变和治疗敏感性。大多数基底型乳腺癌是 TNBC,但也有ERBB2扩增的基底亚型。表观遗传调控因子是细胞状态的关键决定因素,因此,表观遗传突变是肿瘤内异质性的主要来源。编码三甲基化组蛋白 H3 赖氨酸 9 (H3K9me3) 和赖氨酸 36 (H3K36me3) 去甲基化酶的KDM4C在 TNBC 的一个亚群中发生扩增。KDM4C在发育和分化中起着关键作用,在胚胎干细胞 (ESC) 中,它是 OCT4 转录因子的靶标,是 ESC 自我更新和诱导多能干细胞生成所必需的,然而KDM4C在TNBC发生中的作用尚不清楚。
实验设计
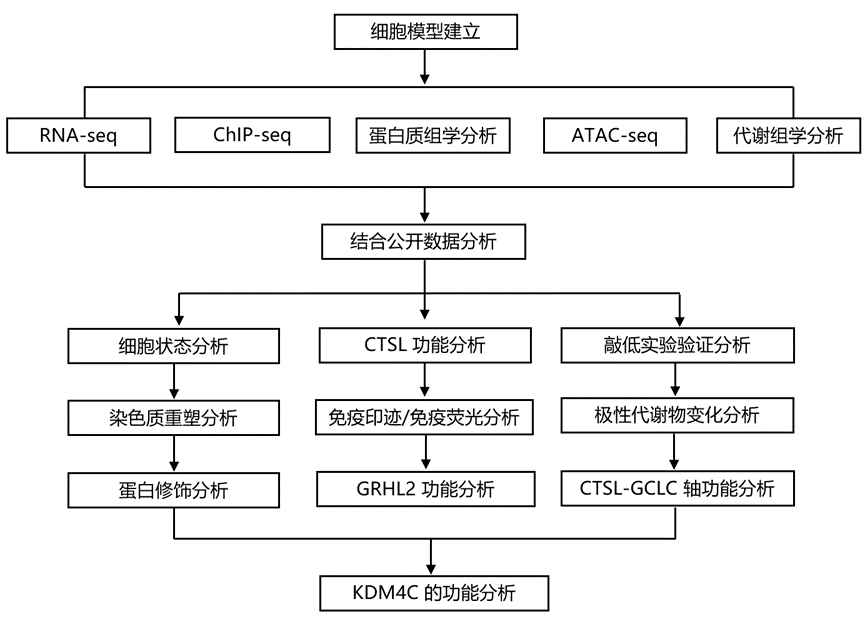
结 果
01

KDM4C在乳腺癌中扩增并促进肿瘤生长
研究团队首先在五种细胞系中使用慢病毒强力霉素 (Dox) 诱导的 shRNA下调KDM4C基因,并通过免疫印迹分析证实了有效的敲低。在所有KDM4C扩增细胞系以及具有中度 KDM4C 表达的非扩增细胞系 (HCC1806) 中,KDM4C 下调均显著降低了体外和体内肿瘤细胞的生长。为了探究KDM4C缺失介导的生长抑制机制,他们首先对四种经Dox诱导表达shKDM4C的基底乳腺癌细胞系进行了转录组分析(RNA-seq),包括KDM4C扩增的细胞(HCC1954和SUM149)和KDM4C未扩增的细胞(HCC1806和HDQP1)。RNA-seq分析证实了KDM4C的有效下调,并且与未扩增的细胞系相比,在KDM4C扩增的细胞系中发现了更显著的转录变化(图1a-b)。利用 Hallmark 标记集通过基因集变异分析 (GSVA) 进行功能注释表明,KDM4C 基因缺失或药物抑制导致多种主要代谢通路(例如胆固醇稳态和氧化磷酸化)活性受到持续抑制,这种情况仅在KDM4C扩增的基底细胞系中出现,但 QC6352 治疗 HDQP1 细胞除外(图1c)。与此观察结果一致,胆固醇稳态和氧化磷酸化是TCGA 队列中KDM4C扩增和KDM4C未扩增的 TNBC 之间差异富集的通路之一,同时还有细胞增殖的关键调节因子(例如 MYC 靶点)(图1d)。他们还在所有四种细胞系中观察到激活的转化生长因子 β (TGF-β) 信号传导和上皮-间质转化 (EMT),表明细胞状态发生了转变。
接下来,他们在四种KDM4C扩增的基础细胞(HCC1954、SUM149、HCC70 和 HCC2157)和两种未扩增的 ER +管腔细胞(T47D 和 MCF7)中对 KDM4C 进行了染色质免疫共沉淀测序(ChIP-seq),发现 KDM4C 染色质峰具有很强的细胞类型特异性,但在启动子和非启动子位点的分布相似。整合 KDM4C 结合和组蛋白修饰模式表明,在大多数细胞系中,KDM4C 与其底物 H3K9me3 和 H3K36me3 互斥,T47D 中的 H3K36me3 除外,并且在所有四种细胞系中都与 H3K4me3 和 H3K27ac 峰共现。他们还评估了 KDM4C 阻断后 H3K9me3 和 H3K36me3 信号强度的整体差异,但 sh KDM4C和 ML324(一种JMJD2 去甲基酶抑制剂)处理均未引起两种KDM4C扩增基底细胞系的实质性变化(图1e)。相反,在 HCC1806 细胞(一种KDM4C非扩增细胞系,具有中等 KDM4C 表达)中 KDM4C 下调后观察到 H3K9me3 和 H3K36me3 的显著差异(图1e)。因此,他们还对 KDM4A 和 KDM4B 进行了 ChIP-seq,发现所有三种 KDM4 酶的基因组结合位点存在明显重叠,并且 KDM4A 结合位点与 ML324 处理后 H3K36me3 水平升高有关。
使用染色质转座酶可及性测序(ATAC-seq)进行分析,抑制KDM4C后,三种细胞系的可及染色质均发生广泛重构,但程度各异。HCC1954和HCC1806细胞主要表现为开放染色质增加,而SUM149细胞的增加幅度较小(图1e)。在KDM4C扩增的细胞中,染色质变化与H3K9me3或H3K36me3信号强度的改变无关,ADARB1基因组位点即为例证(图1f)。此外,在显著获得ATAC信号的区域中,富集的转录因子基序与上皮间质转化(如ZEB1和SMAD2)及抗氧化通路(如BACH1和NF2L1)相关,而CTCF和NFIL3是唯一在缺失ATAC信号的区域中富集的基序(图1g)。
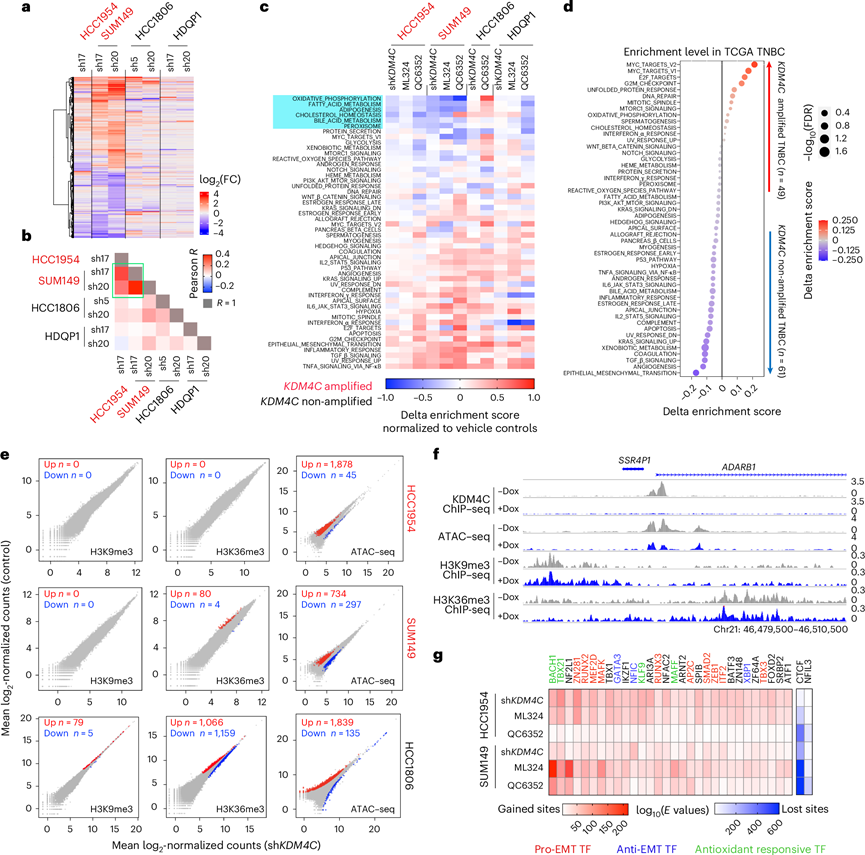
图1. KDM4C 抑制诱导的转录组和染色质重塑。
(a) 热图显示了所有指示的 Dox 诱导的 sh KDM4C细胞模型中载体与 sh KDM4C之间所有 DEG。(b) 热图显示了每次成对比较中所有 DEG 的 log2(FC)的 Pearson 相关性 R 值。(c) 热图描绘了由KDM4C和 KDM4 抑制剂处理下调引起的 50 个 Hallmark 基因特征富集分数的变化。(d) 点图显示TCGA 队列中具有或不具有KDM4C拷贝数增加的 TNBC 之间的 50 个 Hallmark 基因特征富集分数差异。(e) 散点图表示所有 3 种细胞系中对照和 sh KDM4C组之间的 H3K9me3 和 H3K36me3 ChIP-seq(5 kb bin)和合并的 ATAC-seq 峰的log2标准化计数。(f) 具有或不具有 KDM4C 敲低的 SUM149 细胞中ADARB1基因位点处的 KDM4C、ATAC-seq、H3K36me3 和 H3K9me3 信号的基因组轨迹视图。(g) 热图分别显示了在获得和丢失的ATAC位点中前30个和前2个持续且特异性富集的基序。
02
KDM4C 抑制可诱导组蛋白 H3 的蛋白水解裂解
由于KDM4C抑制后出现整体染色质可及性广泛重塑与H3K9me3、H3K36me3修饰改变有限之间的不一致性,他们采用组蛋白质谱(MS)技术进行了全面无偏倚的组蛋白修饰模式分析。出乎意料的是,在ML324处理的SUM149和HCC1954细胞中,检测到组蛋白H3和H4 N端几乎所有修饰标记的丢失,KDM4C敲除也呈现类似但较弱的趋势(图2a)。通常情况下,某些组蛋白修饰水平的下降会因其他修饰的增加所补偿。因此,多数N端修饰的同步丢失可能提示这些组蛋白发生了N端蛋白酶解事件。为验证这一假说,他们对提取的组蛋白进行蛋白质组学分析,发现了与H3A21和H4K16位点非常规切割相吻合的肽段(图2b)。通过定量H3来源的非经典TKAAR肽段,发现HCC1954和SUM149细胞在KDM4C抑制后剪切型H3肽段普遍诱导增加,而T47D细胞变化有限(图2c)。使用C端H3抗体的免疫印迹分析证实,四种KDM4C扩增细胞系在KDM4C敲除或抑制后N端剪切显著增加,而N端H3抗体未检测到明显差异,可能与剪切肽段的快速降解有关(图2d)。KDM4A或KDM4B的下调未诱导组蛋白尾部剪切,且KDM4C诱导的剪切可由野生型而非催化失活突变体KDM4CS198M的异位过表达所阻断,表明该现象具有KDM4C特异性且依赖其去甲基化酶功能。对ML324耐药株的组蛋白质谱分析显示,HCC1954-MLR细胞基线H3剪切水平较高且不受ML324影响,而SUM149-MLR细胞基线剪切较低但ML324诱导效应显著。此外,他们观察到SUM149-MLR细胞中H3Ser10磷酸化肽段显著增加。但SUM149-MLR细胞对H3Ser10激酶(AURKA/AURKB和CDK8)抑制剂未表现差异敏感性,提示该现象可能缺乏功能相关性。
为鉴定介导KDM4C缺失诱导的组蛋白H3/H4尾部剪切的内肽酶,他们在不同蛋白酶抑制剂存在条件下重复质谱分析。通过定量组蛋白H3完整肽段与剪切肽段并结合免疫印迹验证,发现组织蛋白酶L (CTSL)抑制剂(CTSLi-III和SID2668150)或半胱氨酸蛋白酶抑制剂E64d可显著减少H3剪切,而丝氨酸或天冬氨酸蛋白酶抑制剂无此效应(图2e-g),提示CTSL是介导H3剪切的主要候选酶。相比之下,H4肽段的丢失仅由天冬氨酸蛋白酶抑制剂抑肽素A部分挽救,表明H3与H4的蛋白水解机制不同。既往研究报道CTSL在小鼠胚胎干细胞中可在相同位点(H3A21)剪切H3。进一步分析显示,仅在KDM4C扩增细胞系中,KDM4C抑制或下调会增强CTSL活性(图2h-i),这强化了H3剪切与CTSL活性的关联。KDM4抑制导致的CTSL活性增强在SUM149和HCC1954细胞中更为显著,可能与其基线CTSL表达较高有关,且该现象具有KDM4C特异性,因KDM4A或KDM4B敲除未引发类似效应。野生型而非催化失活突变体KDM4C的异位表达可减弱CTSL激活,表明该过程依赖其去甲基化酶功能。
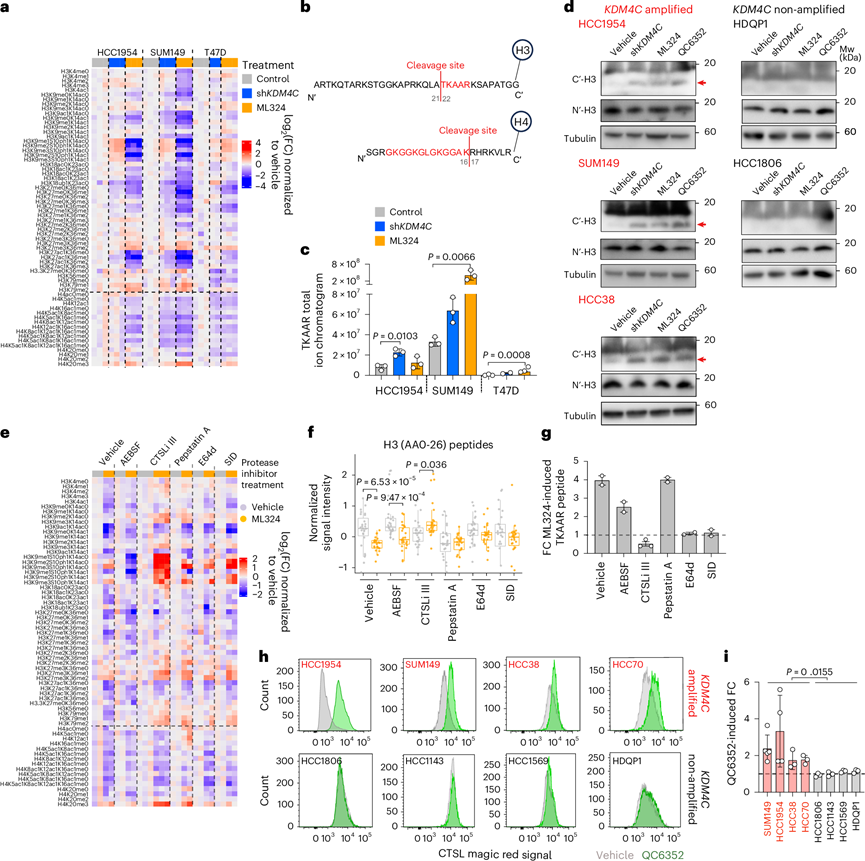
图2. KDM4C 抑制会诱导组蛋白尾的蛋白水解裂解。
(a) 组蛋白肽丰度。(b) KDM4C阻断后组蛋白 H3 和 H4 中的蛋白水解酶裂解位点的示意图。(c) 条形图显示了所示组中剪切的 H3 肽 (TKAAR) 总离子色谱图信号强度。(d)免疫印迹分析。(e) 组蛋白肽丰度。(f) N 端组蛋白 H3(氨基酸位置 0-26)肽丰度的差异。(g) 条形图显示了所示组中 ML324 诱导的截短 H3 肽 (TKAAR) 总离子色谱图信号强度的 FC。(h)流式细胞分析。(i) 条形图总结了 QC6352 诱导的 CTSL 活性中的 FC。
03
CTSL 因 KDM4C 抑制而激活
为了证明 CTSL 在 KDM4C 阻断诱导的组蛋白剪切中的作用,他们使用 CRISPR–Cas9 敲除了五种基底乳腺癌细胞系(三种KDM4C扩增和两种未扩增)中的CTSL。使用分离的细胞裂解物的 CTSL 免疫荧光和免疫印迹表明,它在亲本细胞系中位于核内,但在CTSL敲除(CTSLKO) 系中缺乏核信号。CTSL 的亚细胞定位及其成熟不受 KDM4C 阻断的影响,这表明 KDM4C 缺失诱导的组蛋白裂解不是由于溶酶体破裂诱导的 CTSL 易位到细胞核。使用针对 H3 C 末端的抗体进行免疫印迹分析表明,在所有三种KDM4C扩增细胞系中,CTSLKO系中 KDM4 抑制诱导的 H3 剪切均减少(图3a)。
为了确认 CTSL 定位于染色质,他们在 H3 剪切最明显的 SUM149 细胞中进行了 针对CTSL的ChIP-seq,检测到类似于转录因子结合的尖锐 CTSL 峰,并在 ML324 处理后发生了显著变化(图3b-c)。ML324 处理后消失的 CTSL 峰(有效 CTSL 位点)与 ML324 诱导的差异表达基因 (DEG) 和染色质可及性降低有关(图3d-e),而新增的峰与转录变化的关联有限。ML324 处理后,SUM149-MLR 细胞中的 CTSL 峰强度没有变化,这表明对 ML324 的获得性抗性与组蛋白 H3 尾部剪切减少有关。与未改变的峰相比,ML324 处理后丢失的 CTSL 峰与 KDM4C 结合位点基本重叠(图3c),突出了 CTSL 丢失在 ML324 诱导的染色质重塑中的重要性以及 KDM4C 参与这一过程。
为了进一步证实 KDM4C 抑制后 N 端 H3 的剪切,并排除未知的 N 端 H3 修饰阻碍抗体结合的可能性,他们在 SUM149 细胞中表达了 N 端 GFP 或 V5 标记的 H3,并使用针对 GFP 或 V5 以及 H3 C 端的抗体进行 ChIP-seq。他们检测到在 CTSL 结合位点用 ML324 处理后,使用 GFP 或 V5 抗体的整体 H3 ChIP-seq 信号比使用 C 端 H3 抗体出现更显著的下降,而在随机选择的峰上没有观察到这种下降(图3f-h)。V5 标记的 H3 模型中的改变比 GFP 标记的 H3 模型中的改变更显著,这可能是由于与较大的 GFP 标签相关的空间位阻造成的。
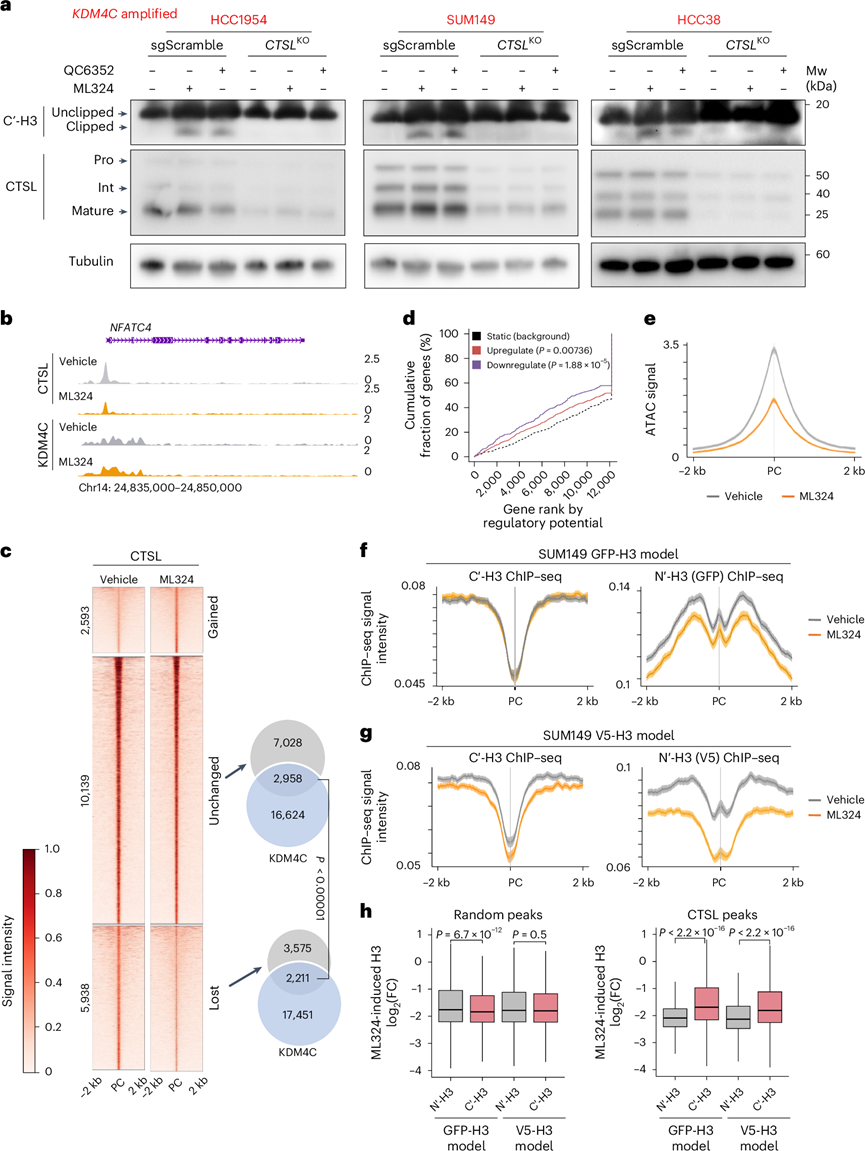
图3. CTSL 是一种由 KDM4C 抑制激活的染色质结合组蛋白 H3 蛋白酶。
(a)免疫印迹分析。(b) 在 NFATC4 基因位点处有或没有 ML324处理的SUM149细胞系中 KDM4C 和 CTSL 结合信号的基因组轨迹视图。(c)热图显示在 SUM149 细胞系中 ML324 处理后差异和未改变的 CTSL 峰。(d) 线图显示结合和表达靶标分析 (BETA),以评估 ML324 处理后 SUM149 细胞中丢失的 CTSL 位点和 DEG 之间的关联。(e-g) 强度图表示 SUM149 细胞系中 ML324 处理后缺失的 CTSL 峰处的 ATAC-seq 信号。(h) 箱线图显示每个指示的 ChIP-seq 样本中 ML324 诱导的 H3 信号变化。
04
GRHL2介导CTSL染色质结合和活性
接下来,他们研究了 CTSL 是如何募集到染色质上的,因为它没有已知的 DNA 结合域。他们对基线 SUM149 细胞中的免疫沉淀 CTSL 进行了 MS 分析(Dox 诱导的 sh KDM4C #17 和 #20,未经 Dox 诱导)。与 IgG 对照相比,CTSL 本身和已知的 CTSL 相互作用蛋白(包括 CTS3 和 CTSB 半胱氨酸蛋白酶抑制剂)是 CTSL 免疫沉淀物中持续富集的最丰富的肽(图4a)。他们检测到了极少数此前未表征的 CTSL 结合蛋白,包括 GRHL2 转录因子和 DEAD-Box 解旋酶。KDM4C的下调并未改变 CTSL 与任何这些蛋白质的相互作用。有趣的是,GRHL2 也是在 HCC1954 和 SUM149 细胞中 CTSL 峰富集的预测最高的转录因子基序(图4b).
他们通过三种蛋白质的共免疫沉淀实验和多色免疫荧光分析证实了 GRHL2、CTSL 和 KDM4C 在细胞核中的结合(图4c)。值得注意的是,使用 CRISPR-Cas9 在 SUM149 细胞中敲除 GRHL2 导致 KDM4C 免疫沉淀物中 CTSL 的缺失,而敲除CTSL 导致 CTSL 免疫沉淀物中 GRHL2 和 KDM4C 的缺失,这排除了观察到的结果是由于 CTSL 抗体与 GRHL2 或 KDM4C 的交叉反应性所致的可能性。
为了验证 CTSL 和 GRHL2 在染色质上的共定位,他们进行了针对GRHL2进行了ChIP-seq,发现在 HCC1954 和 SUM149 细胞系中几乎所有 CTSL 结合位点都与 GRHL2 峰重叠(图4d)。此外, SUM149 细胞中的GRHL2 KO 导致 CTSL 染色质结合几乎完全丧失(图4e)。GRHL2 的缺失也会减弱 KDM4C 抑制诱导的组蛋白 H3 裂解,再次证实了核 CTSL 在此过程中的作用。在 ML324 处理后丢失的 CTSL 位点,GRHL2 峰信号没有受到 ML324 处理的明显影响,证实了 GRHL2 染色质直接结合不依赖于 CTSL。他们还分析了 ML324 处理后 KDM4C 和 GRHL2 峰与丢失的 CTSL 位点之间的结合重叠。虽然 ML324 诱导的丢失的 CTSL 峰(有效 CTSL 位点)中有 38% 与 GRHL2 和 KDM4C 重叠(三重重叠),但这些区域中的 CTSL 峰强度明显低于仅有 CTSL-GRHL2 重叠峰的强度(图4f),这意味着 GRHL2-CTSL 复合物可能具有 KDM4C 独立的功能。三重(KDM4C/GRHL2/CTSL)和双重(GRHL2/CTSL)重叠峰均与 KDM4C 阻断后基因表达抑制密切相关(图4g),并且 DEG 和富集通路显示出相当大的重叠。因此,KDM4C 抑制可能通过直接和间接的方式通过 GRHL2 募集通过 CTSL 介导的组蛋白裂解改变基因表达。
接下来,他们探索了 KDM4C 抑制诱导 CTSL 依赖性组蛋白 H3 剪切的潜在机制。他们假设 GRHL2 或 CTSL 是 KDM4C 的非组蛋白底物,而 KDM4C 抑制会导致其甲基化增强,进而触发 CTSL 介导的组蛋白 H3 剪切。为了验证这一假设,他们首先使用泛赖氨酸甲基化抗体进行免疫沉淀,然后在 KDM4C 敲低或抑制后对 GRHL2 或 CTSL 进行免疫印迹分析。细胞在完全培养基或缺乏蛋氨酸的培养基中生长,以降低细胞内S-腺苷甲硫氨酸 (SAM) 的水平,SAM 是蛋白质甲基化所需的辅助因子。他们在 KDM4C 阻断后在泛赖氨酸甲基化抗体免疫沉淀物中检测到了 GRHL2,并且仅在完全培养基中生长的KDM4C扩增细胞中检测到了 GRHL2(图4h)。为了鉴定GRHL2中特定的甲基化位点,他们对经QC6352处理的SUM149细胞中胶内消化的GRHL2免疫沉淀物进行了MS分析。他们在GHRHL2的K94和K453残基处鉴定出两个赖氨酸单甲基化位点(图4i)。为了确定这些GRHL2赖氨酸甲基化事件的功能相关性,他们外源表达了WT GRHL2或其单或双赖氨酸-精氨酸突变变体(即K94R、K453R和K94R/K453R),这些变体不能发生甲基化,随后使用靶向3′UTR的siRNA下调内源性GRHL2。内源性GRHL2敲低会同时减弱KDM4C抑制诱导的组蛋白H3剪切和CTSL活化,而外源表达野生型GRHL2或GRHL2 K94R突变体可以挽救这种现象(图4j-l)。相反,表达GRHL2 K453R或GRHL2 K94R/K453R双突变体则无法挽救GRHL2缺失的表型(图4j-l)。这些数据表明,GRHL2 K453位点的单甲基化是KDM4C缺失诱导的CTSL活化和组蛋白H3剪切所必需的
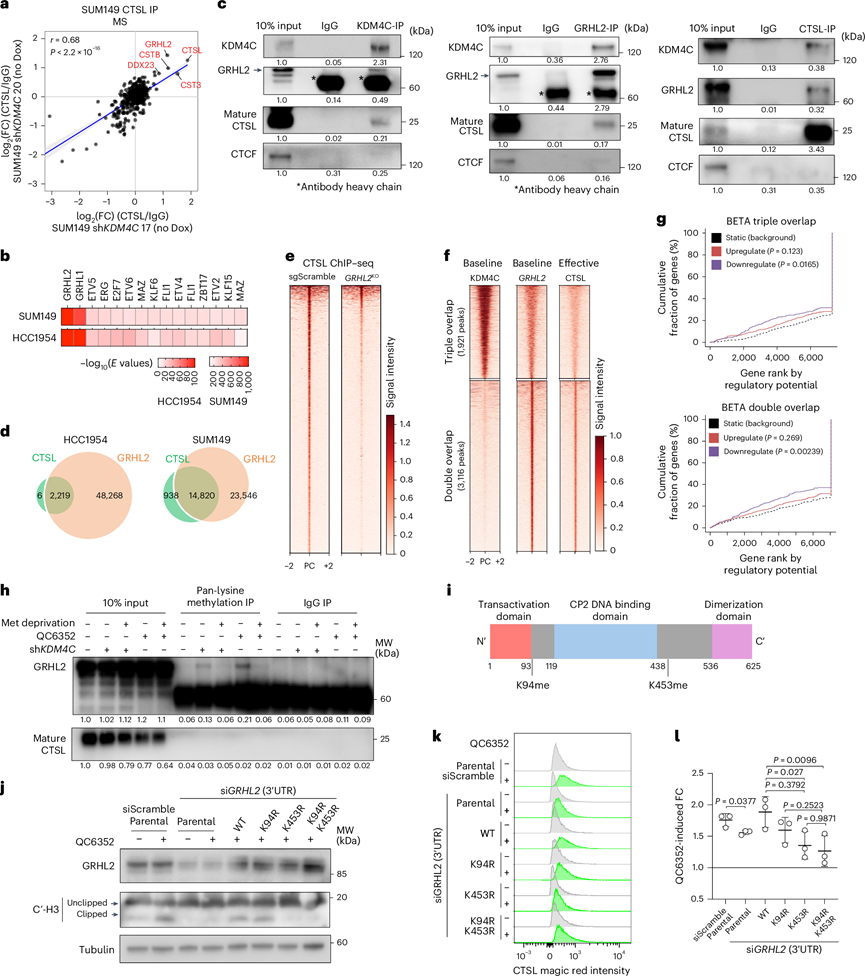
图4. GRHL2 将 CTSL 募集到染色质,其甲基化调节 CTSL 活性。
(a) 散点图显示在未经 Dox 处理的基线时,在 SUM149 细胞模型 sh KDM4C #17 和 sh KDM4C #20 中的 CTSL 免疫沉淀物中检测到的蛋白质的 log 2 (FC) 信号的相关性。(b) 热图描绘了在 SUM149 和 HCC1954细胞系中载体处理细胞的 CTSL 结合位点中富集的转录因子结合位点基序的等级顺序。(c)免疫印迹分析。(d) 维恩图显示 HCC1954 和 SUM149 细胞中 CTSL 和 GRHL2 结合位点的交集。(e) 热图显示乱序对照和GRHL2 KO SUM149 细胞系中 CTSL 染色质结合的总体强度。(f) 热图显示 SUM149 细胞系中的三重(CTSL+ GRHL2+ KDM4C+)和双重(CTSL+ GRHL2+)重叠峰。(g) 线图显示 BETA,用于评估 ML324 处理后 SUM149 中三重和双重重叠峰与差异表达基因的关联。(h)免疫印迹分析。(i) GRHL2 蛋白质结构示意图,显示赖氨酸甲基化位点的位置。(j) 免疫印迹分析。(k)流式细胞分析。(l) QC6352诱导的SUM149细胞模型中CTSL的表达。
05
KDM4C 抑制引起的代谢转变有助于组蛋白剪切
RNA-seq数据显示KDM4C抑制会改变多条代谢通路(图1b),而KDM4C和CTSL的活性均受代谢因素调控。KDM4C是一种依赖氧气和α-酮戊二酸的酶,而CTSL在pH 3.0-6.5且有硫醇化合物存在时活性最佳。为验证"CTSL介导的组蛋白剪切受KDM4C相关代谢变化调控"的假说,他们对四株细胞系(SUM149、HCC1954、HCC70和T47D)及KDM4C抑制前后的SUM149和HCC1954小鼠移植瘤进行了代谢组学分析。248种代谢物的聚类显示,在三株KDM4C扩增的基底型细胞系及移植瘤中(图5a),KDM4C抑制引起广泛的代谢组改变,而在管腔型ER+且无KDM4C扩增的T47D细胞系中仅检测到微小差异(图5a)。对HCC1954和SUM149细胞系及KDM4C抑制处理条件下共同上调代谢物的交集分析显示,次黄嘌呤是唯一重叠的代谢物(图5b),而50种代谢物共同下调,其中还原型谷胱甘肽(GSH)受影响最显著,其他涉及GSH代谢的代谢物也位列其中(图5c)。KDM4C抑制后,还原型(GSH)和氧化型谷胱甘肽(GSSG)及其比值(GSH/GSSG)均下降(图5d),提示GSH生物合成通路抑制导致整体氧化还原失衡。代谢组与转录组变化的联合分析进一步确定,在KDM4C扩增的细胞系和肿瘤中,GSH代谢是与糖酵解、糖异生和戊糖磷酸通路共同下调最显著的通路。KDM4C抑制减弱了整体线粒体呼吸功能(图5e),这与既往关于氧化还原失衡后果的报道一致。
他们进一步研究了KDM4C抑制诱导的GSH水平下降与CTSL介导的组蛋白H3尾部剪切之间的相互作用。与组蛋白剪切和CTSL激活的模式相似,KDM4C抑制或下调在KDM4C扩增的基底型乳腺癌细胞系中引发了更显著的ROS升高(图5f-g)。KDM4C抑制导致的ROS活性增加可通过过表达野生型KDM4C(而非催化失活突变体)得以挽救。通过H2O2直接刺激或谷氨酸-半胱氨酸连接酶抑制剂丁硫氨酸亚砜胺(BSO)阻断GSH生物合成来提升ROS水平,可增加组蛋白剪切和CTSL活性,且在同一细胞中检测到高ROS和活性CTSL。相反,使用细胞可渗透型GSH衍生物GSH乙酯(GSE-EE)中和ROS,能有效降低SUM149细胞的CTSL活性(图5h)。GSH-EE处理还减少了QC6352诱导的SUM149细胞组蛋白H3剪切(图5i)。他们同时检测了KDM4C抑制或GSE-EE处理是否会改变CTSL成熟过程。免疫印迹分析显示,细胞核与细胞质中成熟CTSL比例在各处理组间无显著差异,表明观察到的CTSL活性变化并非由CTSL成熟过程改变所致。
鉴于ROS和KDM4C-GRHL2相互作用均能触发CTSL活化和组蛋白尾部剪切,他们进行了时间进程实验以确定这些事件的时间顺序。在KDM4C抑制剂治疗的6天内,ROS和CTSL活性均呈逐渐升高的趋势,但CTSL活化早在治疗后24小时就已发生,而ROS水平则在36至60小时之间开始升高(图5j)。这些数据表明,KDM4C抑制后GRHL2 K453甲基化的增加可能是CTSL活化的初始触发因素,而GSH抑制和ROS诱导则是下游事件,它们通过正反馈进一步增强CTSL活性。
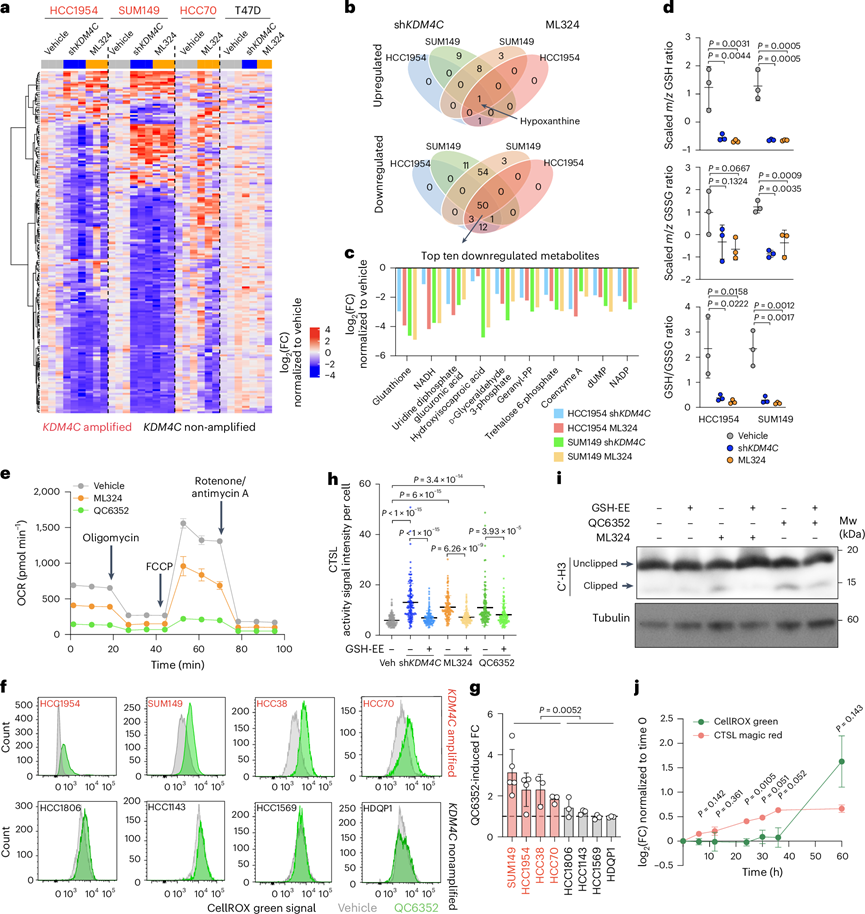
图5. KDM4C 阻断导致氧化还原失衡,从而激活 CTSL。
(a) 248 种极性代谢物的聚类情况。(b) 维恩图显示了在用 Dox(sh KDM4C )或 ML324 处理后,表达 sh KDM4C的 HCC1954 和 SUM149 细胞中上调或下调代谢物的交集。(c) 条形图表示使用 Dox 或 ML324 处理的表达sh KDM4C的 HCC1954 和 SUM149 细胞中持续下降的前十种代谢物。(d) 点图描绘了使用 sh KDM4C或 ML324 处理的 HCC1954 和 SUM149 细胞系中标准化的还原(GSH)和氧化(GSSG)GSH 水平及其比率。(e)线图描述了用 DMSO、10 μm ML324 或 1 μm QC6352 处理 3 天的 SUM149 细胞系中海马线粒体应激分析记录的氧消耗率(OCR)变化。(f)流式细胞分析。(g) 条形图显示了QC6352诱导的CellROX绿信号中的FC。(h) 点图描绘了从3幅代表性荧光图像中的120个单个细胞定量CTSL活性信号。(i)免疫印迹分析。(j) 线图显示了QC6352在SUM149细胞中在指定时间点诱导的CTSL和CellROX信号。
06
KDM4C 抑制会降低 GCLC,导致氧化还原失衡
为深入探究KDM4C抑制导致GSH下降的机制,他们通过RNA-seq数据比较了GSH生物合成通路关键酶及转运体的表达水平。研究发现多个基因表达下调,包括GSS(编码谷胱甘肽合成酶)、GCLC和GCLM(分别编码谷氨酸-半胱氨酸连接酶催化亚基与调节亚基)。免疫印迹分析证实,在KDM4C下调或抑制后,HCC1954和SUM149细胞系中限速酶GCLC均呈现显著且一致的表达降低(图6a)。通过免疫荧光在SUM149移植瘤模型中进一步验证了Dox诱导的KDM4C敲除可导致GCLC减少(图6b)。GCLC作为GSH合成的首个限速酶亚基,负责催化谷氨酸与半胱氨酸缩合生成GSH前体γ-谷氨酰半胱氨酸。他们对34株TNBC细胞系的转录组与代谢组分析发现,GCLC与GSH水平呈正相关,且GSH是驱动TNBC代谢异质性的关键因素,可将样本分为高、低两组。此外,TCGA数据库中基底型乳腺癌样本的KDM4C与GCLC mRNA水平呈显著正相关(图6c),提示二者在临床样本中存在协同调控关系。
他们进一步探究了KDM4C调控GCLC表达的机制。首先,外源表达野生型(而非催化失活突变体)KDM4C能逆转KDM4C敲除导致的GCLC下调,证实该现象具有KDM4C特异性。ChIP-seq数据显示,在SUM149和HCC1954细胞中,CTSL、GRHL2和KDM4C的峰均重叠出现在GCLC启动子区域,且KDM4C抑制显著降低该基因组区域的染色质可及性(图6d),提示GCLC下调可能是CTSL介导的组蛋白H3尾部剪切的结果。这一发现在CTSLKO细胞中得到验证——KDM4C抑制未能降低GCLC表达。在观察到CTSL介导组蛋白H3剪切的KDM4C扩增模型中,CTSL缺失还消除了KDM4C抑制诱导的肿瘤生长抑制效应(图6e-g)。RNA-seq分析显示,SUM149 CTSL敲除细胞中QC6352诱导的转录组改变较弱。值得注意的是,CTSL敲除能部分恢复sgScramble对照组细胞中受QC6352调控的基因特征,包括氧化磷酸化和脂肪酸代谢等关键代谢功能(图6h)。与此一致,KDM4C扩增的CTSLKO细胞中,KDM4C抑制相关的ROS升高得到部分缓解(图6i-j)。最后,在CTSLKO的SUM149细胞移植瘤中,基线GSH水平低于对照组,KDM4C抑制仍未能降低GSH水平(图6k),而在无KDM4C扩增的HCC1806细胞移植瘤中未检测到GSH水平变化(图6k)。这些结果确定了 CTSL-GCLC 轴是KDM4C扩增基底乳腺癌中 KDM4C 缺失相关代谢组学和表观遗传重塑以及肿瘤生长抑制的关键介质(图6l)。
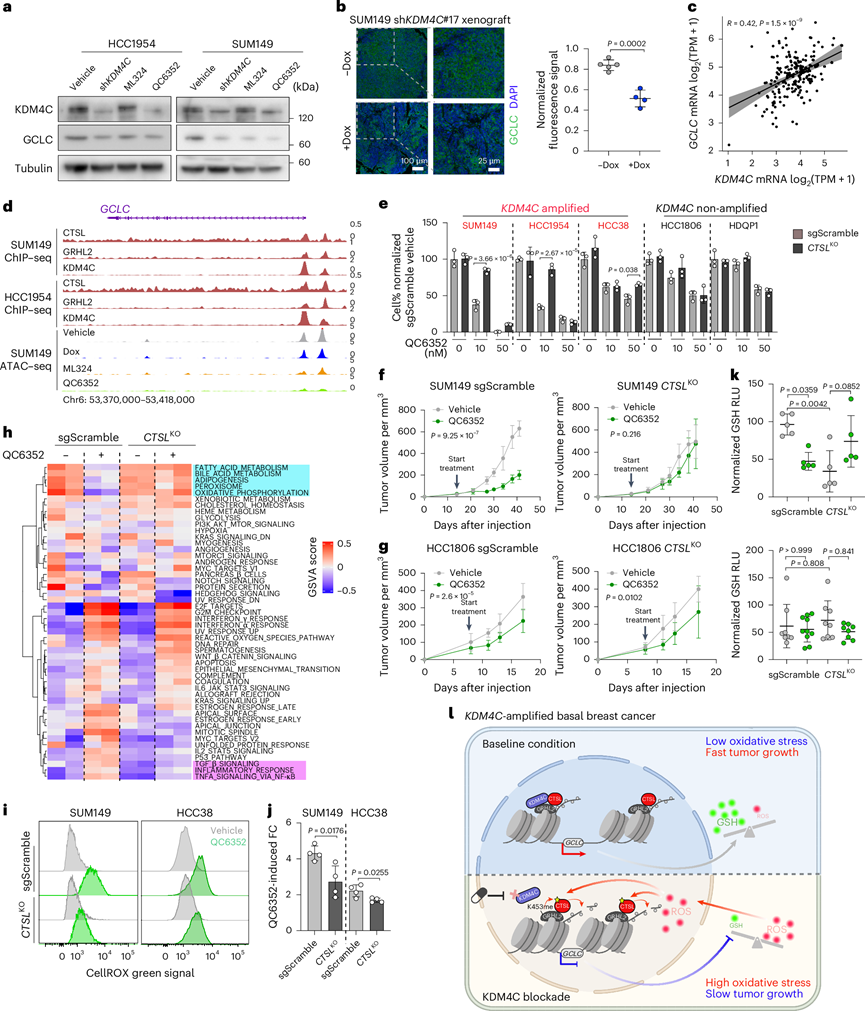
图6. KDM4C 阻断通过 CTSL降低GCLC表达。
(a)免疫印迹分析。(b) GCLC 免疫荧光染色代表性图像。(c) 散点图描绘了TCGA 队列中 190 个乳腺基底肿瘤中KDM4C和GCLC mRNA 水平之间的相关性。(d) HCC1954 和 SUM149 细胞在GCLC基因组位点的 CTSL、GRHL2 和 KDM4C 结合的基因组轨迹视图。(e) 条形图显示了在所示组中用 DMSO 处理的 sgScramble 细胞模型标准化的细胞百分比。(f-g) 异种移植瘤的肿瘤体积。(h) 热图显示基于 50 个标志性基因特征的 GSVA 富集分数对样本进行无监督聚类。(i)流式细胞分析。(j) 条形图描绘了合并三个独立实验的 QC6352 诱导的 CellROX信号强度FC。(k) 点图描绘了在终点收集的 SUM149 和 HCC1806 异种移植瘤中 GSH 水平标准化为肿瘤重量。(l)功能示意图。
+ + + + + + + + + + +
结 论
本研究发现了KDM4C在KDM4C扩增的基底型乳腺癌中的独特作用。KDM4C抑制会重塑染色质和转录组,而不会显著改变其典型底物——H3K9me3和H3K36me3。相反,KDM4C缺失会导致由组织蛋白酶L (CTSL) 介导的组蛋白H3蛋白水解性裂解,从而导致谷氨酸-半胱氨酸连接酶表达下降和活性氧增加。CTSL由GRHL2 转录因子募集到染色质上,该转录因子在KDM4C抑制后在赖氨酸453位点发生甲基化,从而触发CTSL组蛋白剪切活性。CTSL的缺失挽救了KDM4缺失介导的肿瘤抑制。本研究揭示了KDM4C连接细胞氧化还原调控和染色质重塑的功能。
+ + + + +



