

 English
English文献解读|Signal Transduct Target Ther(52.7):ENO2驱动肿瘤细胞诱导的M2巨噬细胞极化,从而促进结直肠癌肝转移
✦ +
+
论文ID
原名:ENO2 drives tumor cell-induced M2 macrophage polarization to promote colorectal cancer liver metastasis
译名:ENO2驱动肿瘤细胞诱导的M2巨噬细胞极化,从而促进结直肠癌肝转移
期刊:Signal Transduction and Targeted Therapy
影响因子:52.7
发表时间:2026.05.05
DOI号:10.1038/s41392-026-02732-2
背 景
结直肠癌(CRC)是全球第三大常见恶性肿瘤,每年新增病例超过190万例,死亡主要由远处转移引起。在转移部位中,肝脏是最常见且最致命的,占CRC相关死亡病例的70%以上。目前的一线全身治疗,包括抗表皮生长因子受体(EGFR)和抗血管内皮生长因子(VEGF)靶向药物,对结直肠癌肝转移(CRLM)患者的生存获益有限,尤其是对于绝大多数微卫星稳定型(MSS)CRC患者,这类患者对单药免疫检查点阻断疗法存在原发性耐药。虽然在手术切除、全身化疗和新兴免疫疗法等多模式治疗方面取得了进展,但CRLM的5年生存率仍然低于20%。这种严峻的预后凸显了迫切需要破译肝脏趋向性和免疫逃逸的分子驱动因素,这是实现转移性生长的关键生物学特征。
实验设计
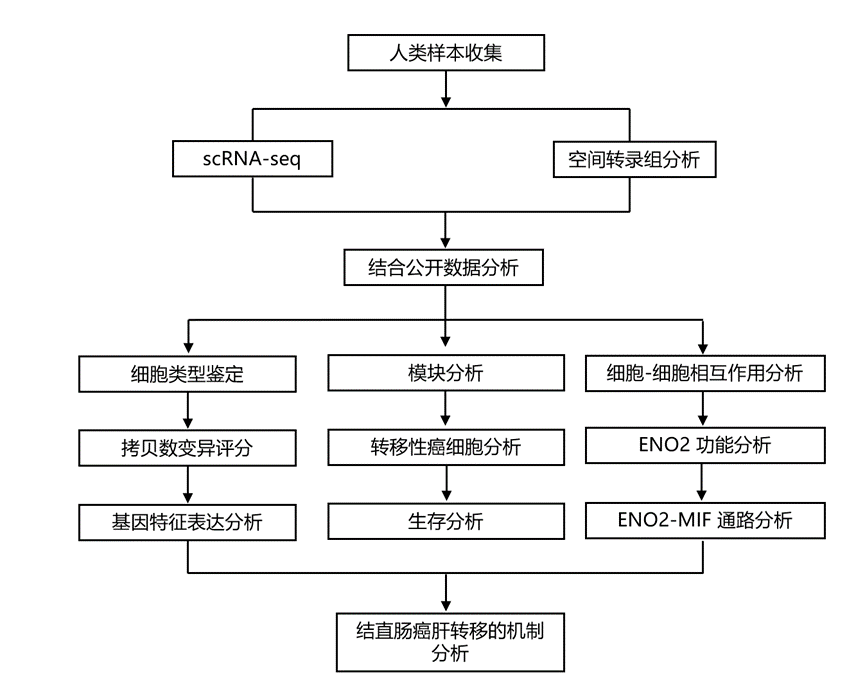
结 果
01

单细胞图谱揭示肝转移病灶恶性程度增强
为了阐明结CRLM的细胞生态系统,研究团队对来自6例患者的15份组织标本进行了单细胞RNA测序(scRNA-seq):其中3例为非转移性结直肠癌(CRC-nM),3例为CRLM。对于CRLM患者,配对样本包括原发性结直肠肿瘤(CRC)、邻近肠道组织和肝转移灶(LM)。
在三例CRLM患者中,无监督聚类分析显示,上皮细胞是原发灶和转移灶肿瘤组织中的主要细胞成分(图1b-d)。研究团队进一步对上皮细胞进行亚聚类分析,发现肿瘤组织中癌细胞的富集程度高于邻近组织(图1e-f)。值得注意的是,与原发灶癌细胞相比,肝转移灶癌细胞表现出多方面的恶性增强证据。基因组不稳定性升高表现为拷贝数变异(CNV)评分显著升高(图1g),同时伴有增殖驱动基因和缺氧反应基因的转录上调(图1h)。这些分子改变最终导致功能增强的表型,转移灶细胞表现出显著增强的增殖能力和缺氧适应能力(图1i-j)。总的来说,单细胞图谱描绘了CRLM的细胞生态系统,并揭示了肝转移病灶的恶性程度增加,与原发肿瘤相比,其特征是基因组不稳定性更高、增殖能力更强、缺氧适应性更强。
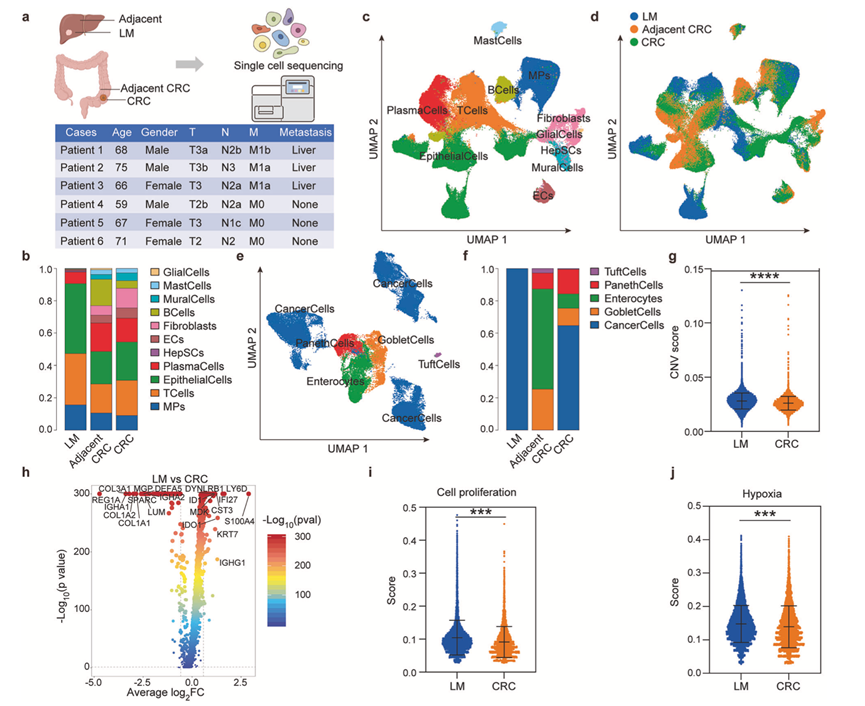
图1. 单细胞图谱揭示肝转移灶恶性程度增强。
(a) 研究设计:对3例非转移性结直肠癌(CRC)患者和3例结直肠癌肝转移(CRLM)患者的组织进行单细胞RNA测序(scRNA-seq)。(b) 3例CRLM患者肝转移灶(LM)、邻近组织(CRC邻近组织)和原发性CRC组织中主要细胞类型的组成。(c) 11种细胞类型的UMAP可视化图,这些细胞类型由标记基因注释。(d) 注释细胞的组织来源分布。(e) 上皮细胞亚群分析显示肿瘤中癌细胞富集。(f) 上皮细胞亚群的比例丰度。(g) 原发性(CRC)和转移性(LM)部位癌细胞的拷贝数变异(CNV)评分。(h) 火山图显示scRNA-seq数据中差异表达的基因,比较肝转移灶(LM)细胞和原发性CRC细胞。(i) 转移性癌细胞与原发性癌细胞的细胞增殖特征。(j) 肝转移中缺氧相关基因表达。
02
基于模块的转移启动型癌细胞亚群发现
为了揭示转移启动的癌细胞亚群,他们对scRNA-seq数据进行了基于模块的分析。利用均匀流形逼近和投影(UMAP)方法对肿瘤细胞进行降维,结果显示转移灶和原发灶中均存在八个不同的细胞亚群(图2a-b),这些亚群表现出不同程度的恶性程度。
接下来,他们应用模块分析模型对癌细胞进行聚类,并将结果与上述转移相关亚组进行比较。在“模块11”中得分阳性的细胞主要富集于癌细胞亚组1、2和3中,这些亚组共同表现出转移性癌细胞的特征(图2d)。重要的是,根据多因素Cox回归模型(图2e),“模块11”中的几个基因与癌症基因组图谱(TCGA)结肠腺癌(COAD)数据集中的CRC患者预后不良显著相关。
他们进一步分析了转移性和非转移性患者原位病灶的单细胞谱。聚焦于原发病灶,UMAP降维分析识别出六个亚组,其分布在转移性和非转移性患者之间存在差异(图2f-g)。对这些亚组的经典恶性特征进行评估发现,癌细胞亚组3表现出显著的上皮-间质转化(EMT)特征,提示其可能在启动转移中发挥作用(图2h-i)。综上所述,这些结果表明“模块11”基因集符合转移性CRC细胞的表达特征,并明确指出具有EMT特征的独特癌细胞亚群(亚组3),该亚群可能代表原发肿瘤中一种转移启动状态。
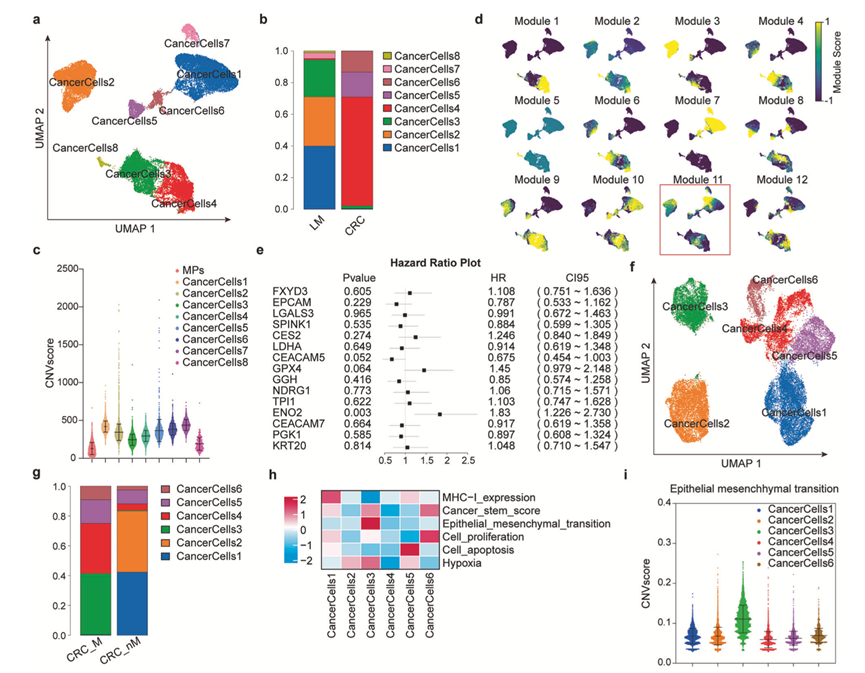
图2. 基于模块的转移启动型癌细胞亚群发现。
(a-b) 对肿瘤细胞进行UMAP降维分析,并展示了转移灶和原发灶中八个机械亚群的分布。(c) 通过CNV评分评估八个癌细胞亚群的恶性程度,以突变负荷较低的巨噬细胞(MP)作为阴性对照。(d) 基于模块分析模型对癌细胞进行聚类和降维。(e) 使用TCGA-COAD数据集中的多因素Cox回归模型分析“模块11”基因集中的基因与结直肠癌预后的相关性。(f-g) 对来自 6 个原发病灶组织的肿瘤细胞进行了UMAP 降维分析。(h-i) 利用肿瘤细胞的典型恶性特征分析了六个肿瘤细胞亚群的潜在特征。
03
ENO2驱动转移进展,并可作为治疗靶点
基于转移相关特征的鉴定,他们从“模块11”和癌细胞3的表达谱中筛选出10个候选基因(图3a)。其中,只有烯醇化酶2(ENO2)证实为重要的预后决定因素,其高表达与CRC患者生存期显著缩短相关(图3b)。ENO2是一种糖酵解酶,催化2-磷酸甘油酸转化为磷酸烯醇式丙酮酸,正是这种双重性的一个例证。除了其在三磷酸腺苷 (ATP) 生成中的经典作用外,ENO2 在神经内分泌肿瘤、肺癌和CRC中均存在异常表达,并且与化疗耐药性相关。从机制上讲,ENO2 可通过核转位和组蛋白去乙酰化酶 (HDAC) 抑制来调节基因转录。ENO2 还可通过蛋白质-蛋白质相互作用稳定致癌激酶。然而,其作为CRLM中免疫微环境相互作用的关键调节因子的潜力尚未得到充分研究。南京和广州队列的临床验证证实,与正常组织相比,原发肿瘤和肝转移灶中ENO2表达均上调(图3c),并证实其与患者生存期呈负相关(图3d)。使用GSE41258数据集和TNMplot在线工具验证了ENO2表达与CRLM之间的相关性。在C57BL/6J小鼠模型中进行的功能性研究揭示了ENO2敲除对肿瘤转移的显著抑制作用。皮下异种移植瘤实验表明,ENO2敲除组的肿瘤生长显著减弱,表现为肿瘤体积和重量的降低(图3e)。更重要的是,脾脏注射模型显示,ENO2缺陷细胞的肝脏转移受到抑制,转移结节减少了50%以上(图3f)。
药理学抑制ENO2(ENOblock)进一步强调了其治疗意义,该抑制剂能有效阻断ENO2的促转移功能(图3g)。多重免疫荧光证实了ENO2与EMT诱导之间的机制联系,显示ENO2表达肿瘤中E-钙黏蛋白丢失和波形蛋白增加(图3h)。这表明靶向ENO2具有一定的临床应用前景。
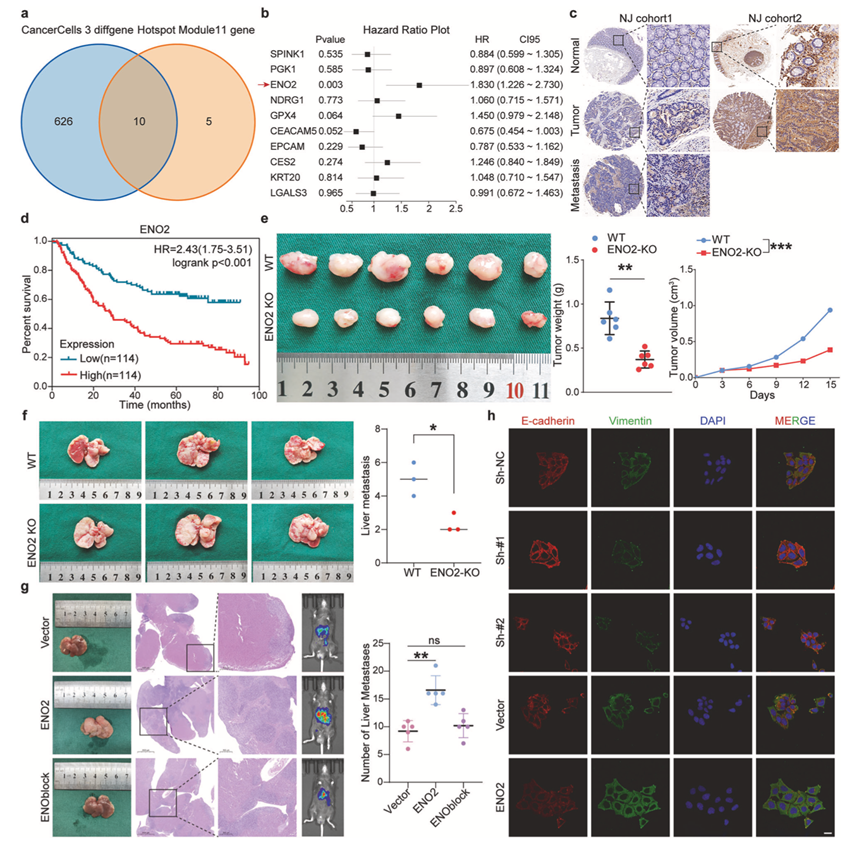
图3. ENO2驱动转移进展并可作为治疗靶点。
(a) 从“模块11”基因集和“癌细胞3”特征基因中筛选出10个候选基因。(b) 基于TCGA-COAD数据库,采用多因素Cox回归分析这10个候选基因与结直肠癌预后的相关性。(c) 在多个独立队列中,采用免疫组化方法检测ENO2表达与结直肠癌进展的相关性。(d) 利用来自南京和广州的三个独立样本队列,进行Kaplan- Meier生存分析,探讨ENO2表达与结直肠癌预后的关系。(e) 在C57BL/6J小鼠皮下肿瘤模型中观察ENO2敲除DLD-1细胞的致瘤能力,并比较肿瘤组织的重量和体积。(f) 利用C57BL/6J小鼠脾内注射模型,观察了ENO2敲除DLD-1细胞与野生型细胞形成的肝转移结节数量。(g) 使用ENO2药理学抑制剂(ENOblock)验证靶向ENO2抑制结直肠癌肝转移(CRLM)的潜力。(h) 对对照组和ENO2调控的癌细胞进行EMT标志物(E-钙黏蛋白、波形蛋白)的多重免疫荧光染色。
04
ENO2 通过与 MIF 的直接相互作用来调控 M2 巨噬细胞极化
为了研究癌细胞如何塑造肿瘤微环境(TME),他们利用CellChat分析了细胞间通讯。分析结果显示,ENO2+癌细胞是TME中的主要信号枢纽,其中巨噬细胞迁移抑制因子(MIF)信号通路表现出特别强且特异性的外向/内向信号(图4a)。聚焦于MIF介导的相互作用,ENO2+癌细胞与巨噬细胞的通讯显著增强,而ENO2-癌细胞则不然(图4b-c)。这一计算预测结果与观察结果相符,即转移灶中的巨噬细胞比原发性或非转移性肿瘤中的巨噬细胞表现出更高的M2极化倾向(图4d)。
为了阐明其机制基础,他们通过共免疫沉淀(Co-IP)结合质谱分析鉴定了ENO2和MIF之间的直接物理结合(图4e-f)。体外谷胱甘肽S-转移酶(GST)下拉实验(使用纯化的重组蛋白)证实了这种相互作用是直接的(图4g)。为了确定分子界面,他们进行了分子对接模拟,预测了ENO2和MIF之间的特异性结合模式(图4h)。随后,他们使用两种蛋白的结构域特异性突变体进行了Co-IP实验,验证了这些预测,结果表明破坏预测的界面完全消除了它们的结合(图4i)。这些结果证实了癌细胞中 ENO2 和 MIF 之间存在直接的物理相互作用,并表明这种相互作用是 ENO2 +癌细胞向巨噬细胞增强 MIF 介导信号传导的基础,从而促进了 M2 极化的免疫抑制微环境。
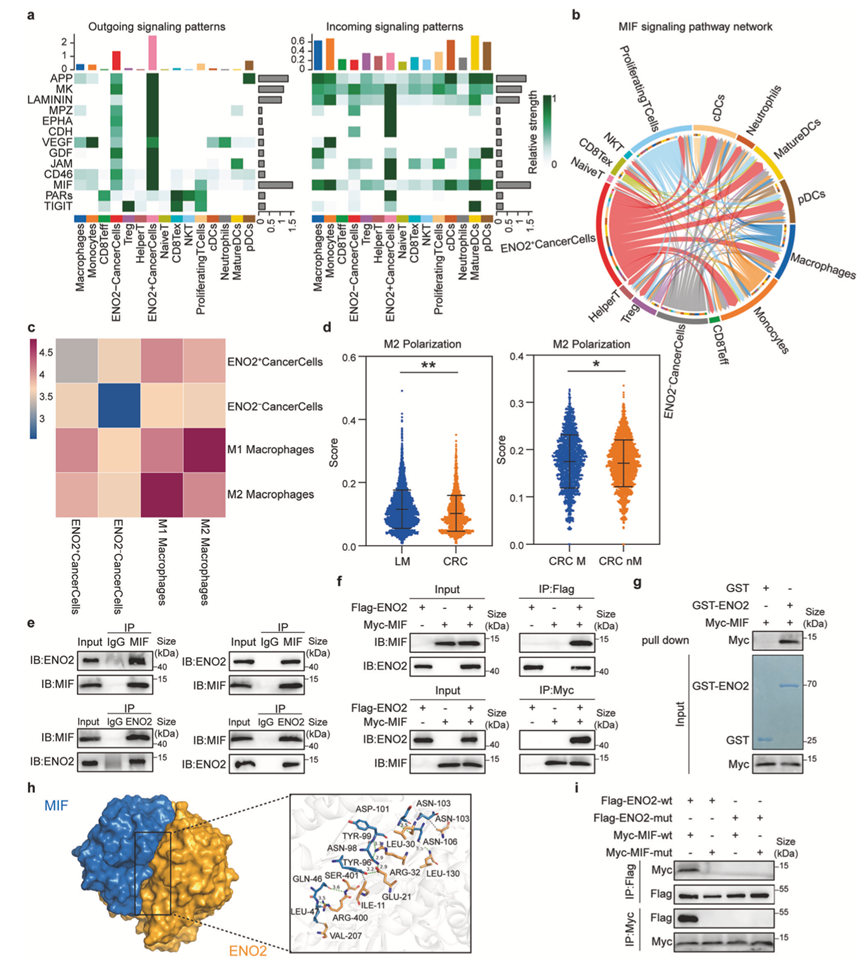
图4. ENO2通过与MIF的直接相互作用调控M2巨噬细胞极化。
(a) 基于CellChat分析不同细胞类型参与微环境信号通讯的强度和潜在方式。(b-c) 在MIF信号通路参与的情况下,分析了ENO2-/-癌细胞与不同细胞类型之间的相互作用强度。(d) 与原位或非转移性原位病变相比,转移性病变和转移性原位病变中的巨噬细胞更倾向于M2极化。(e) 免疫共沉淀(Co-IP)检测证实了内源性ENO2与MIF之间的相互作用。(f) 对DLD-1细胞中内源性MIF和ENO2进行共免疫沉淀分析。(g) GST下拉实验表明MIF和ENO2在体外存在直接相互作用。(h-i) 分子对接模拟预测了ENO2和MIF之间的关键结合界面,并使用结构域特异性突变体进行相互免疫沉淀分析。
05
ENO2通过抑制泛素介导的降解来稳定MIF,从而激活促转移信号通路
接下来,他们研究了ENO2-MIF相互作用的功能后果。邻位连接分析(PLA)证实了癌细胞内源性ENO2-MIF相互作用的存在(图5a)。功能上,在DLD-1细胞中敲低ENO2导致MIF蛋白水平显著降低,而在HCT-116细胞中过表达ENO2则增加了MIF的丰度,表明ENO2正向调控MIF的稳态水平(图5b)。
鉴于已知MIF受泛素介导的蛋白酶体降解调控,他们探究了ENO2是否通过干扰该过程来稳定MIF。使用蛋白酶体抑制剂MG132处理后,ENO2敲低细胞中的MIF水平得以恢复,而ENO2过表达细胞中的MIF水平则进一步升高(图5c)。在MG132处理的细胞中,ENO2敲低显著增强了MIF的多聚泛素化,而ENO2过表达则产生了相反的效果(图5d)。
结构域定位实验证实,这种稳定性需要完整的结合界面,因为ENO2催化结构域突变体无法保护MIF免受泛素化修饰(图5e-f)。最后,他们研究了其对下游致癌信号通路的影响。ENO2表达的调控相应地改变了STAT3和NF-κB亚基P65的磷酸化水平(图5g),这两个关键通路均由MIF激活。这些数据表明,ENO2通过拮抗CHIP介导的多聚泛素化及其后续的蛋白酶体降解来结合并稳定MIF蛋白,从而增强癌细胞中MIF依赖的STAT3和NF-κB信号通路的激活。
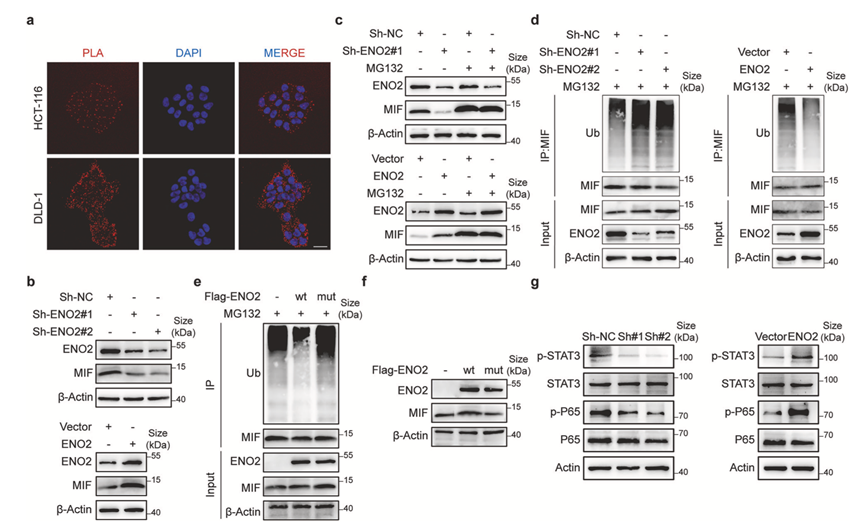
图5. ENO2 通过抑制泛素介导的降解来稳定 MIF。
(a) 邻位连接分析 (PLA) 显示细胞内源性 ENO2 与 MIF 之间的相互作用。(b-g) Western blot 分析。
06
ENO2诱导M2巨噬细胞极化,从而促进肝转移
为了探究ENO2-MIF轴是否在功能上重塑肿瘤免疫微环境,他们首先分析了二者的空间关系。空间转录组分析(ST)显示,在转移灶中,ENO2阳性癌细胞与M2样巨噬细胞存在显著的共定位(图6a)。对多个ST样本进行定量相关性分析证实,这两个细胞群之间存在一致且显著的正相关性。这种空间关联促使他们进一步探究表达ENO2的癌细胞是否能够直接诱导M2极化。
利用体外共培养系统,他们发现与ENO2敲低癌细胞共培养的巨噬细胞表现出M1标志物(CD86)增加和M2标志物(CD206)减少,而与ENO2过表达细胞共培养的巨噬细胞则呈现相反的趋势(图6b)。这种ENO2依赖的M2极化在体内也得到了重现,表现为与对照组相比,ENO2过表达细胞来源的异种移植瘤中CD206+巨噬细胞的比例更高(图6c)。使用患者来源的类器官(PDO)进一步证实了癌细胞内源性ENO2的关键作用;与 ENO2 敲除 PDO 共培养的巨噬细胞的 M2 表型降低,而与 ENO2 过表达 PDO 共培养的巨噬细胞则表现出增强的 M2 极化(图6d)。
随后,他们评估了该轴对体内转移的功能性影响。在C57BL/6J小鼠脾移植-肝转移模型中,癌细胞中ENO2的过表达显著促进了肝转移的形成(图6e-f)。重要的是,这种促转移作用可由MIF抑制剂ISO-1或M2极化抑制剂LPS消除。相反,ENO2敲低导致的转移减弱可通过给予M2激动剂IL-4或在癌细胞中强制过表达MIF来恢复(图6e-f),免疫组织化学证实了各组间M2极化的丰富程度。这些结果表明,ENO2阳性癌细胞通过分泌MIF驱动肿瘤微环境中M2巨噬细胞的极化,进而促进肝转移的形成。
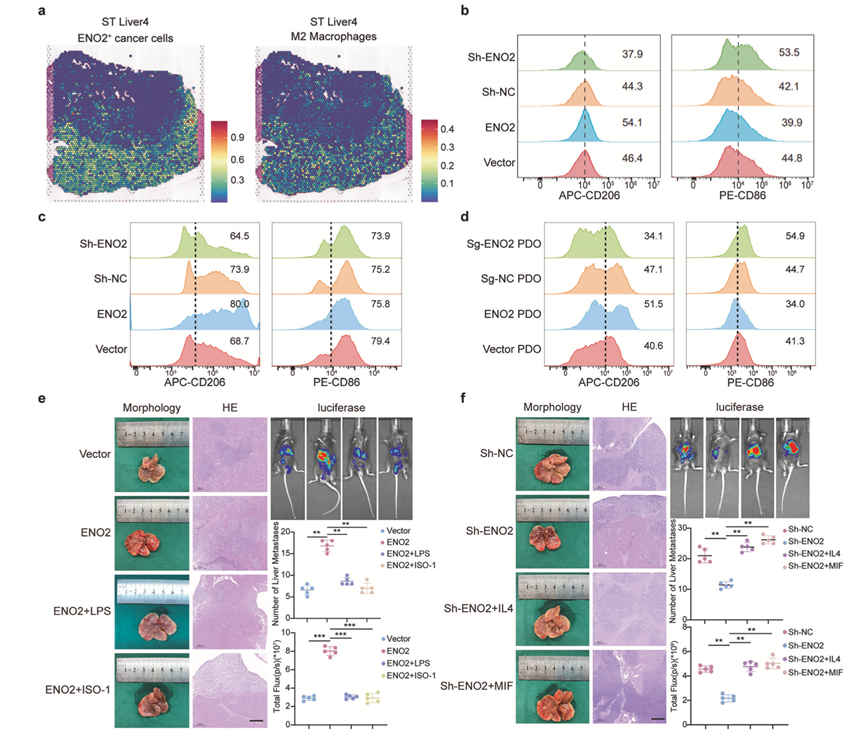
图6. ENO2诱导M2巨噬细胞极化驱动肝转移。
(a) 空间特征图显示了ST_liver4中ENO2+癌细胞和M2巨噬细胞的反卷积比例。(b-c) 流式细胞分析共培养系统和异种移植瘤中巨噬细胞标志物CD86和CD206的表达。(d) 代表性流式细胞图显示了与患者来源类器官(PDO)模型共培养的巨噬细胞上CD206和CD86的表达。(e-f) 利用C57BL/6J小鼠脾脏移植模型,研究ENO2表达对肝转移形成的影响。
07
NO2 募集 HSP90 以拮抗 CHIP 介导的 MIF 泛素化和降解
为了进一步阐明ENO2如何稳定MIF,他们试图鉴定其分子互作蛋白。质谱分析ENO2复合物发现分子伴侣热休克蛋白90 (HSP90) 是其结合蛋白(图7a),这一结果通过内源性蛋白的双向免疫共沉淀(Co-IP)实验得到证实(图7b-c)。有趣的是,Co-IP实验表明ENO2增强了HSP90与MIF之间的物理结合,提示ENO2可能作为支架蛋白募集HSP90至MIF(图7d)。
由于已知E3连接酶CHIP靶向HSP90底物蛋白进行降解,他们假设ENO2可能通过拮抗CHIP来保护MIF。与此一致,ENO2敲低导致的MIF水平降低可通过同时敲低CHIP而恢复(图7e),而ENO2过表达诱导的MIF积累则可通过共表达CHIP而抑制(图7f)。从机制上讲,ENO2敲低显著增强了CHIP介导的MIF多聚泛素化(图7g)。
为了确定泛素化位点,他们构建了MIF突变体,其中候选赖氨酸残基(K32、K66、K77)被替换。泛素化实验表明,K66位点的突变(而非K32或K77位点的突变)显著抑制了CHIP介导的MIF多聚泛素化(图7h-i),从而确定K66为主要泛素化位点。综上所述,这些结果表明ENO2募集HSP90至MIF,形成三元复合物,该复合物竞争性抑制E3泛素连接酶CHIP,从而抑制CHIP介导的MIF在K66位点的泛素化及其后续的蛋白酶体降解。
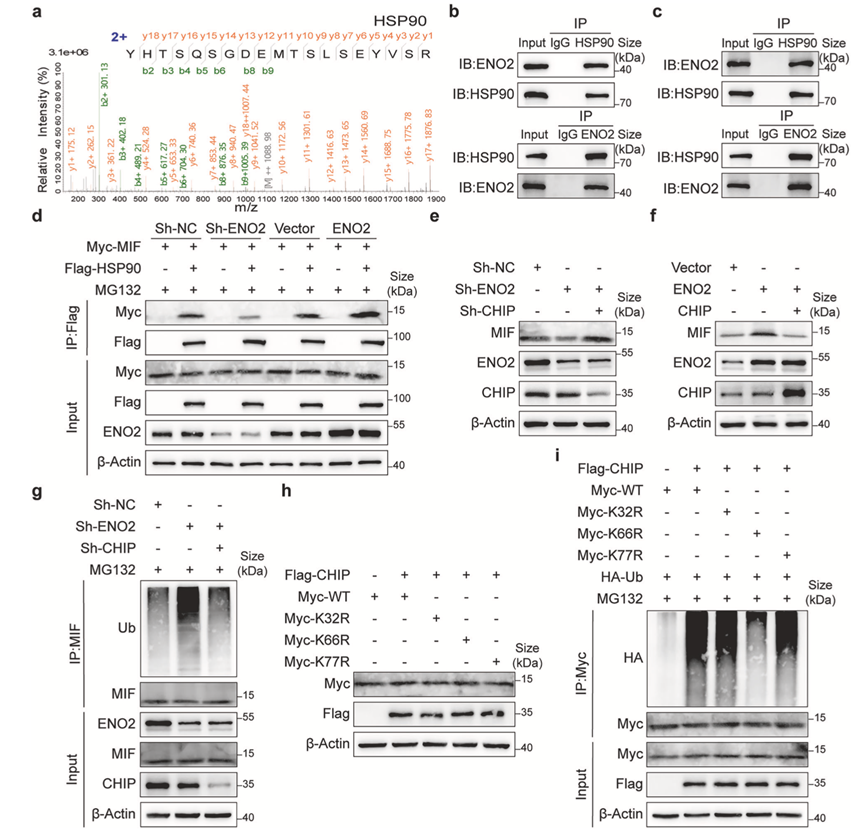
图7. ENO2 募集 HSP90 以拮抗 CHIP 介导的 MIF 泛素化和降解。
(a) 代表性的液相色谱-串联质谱 (LC-MS/MS) 图谱,鉴定出 HSP90 为 ENO2 相互作用蛋白,并突出显示了匹配的肽段片段。(b-i) 免疫沉淀和Western blot 分析。
05
吡硫辛作为ENO2-MIF相互作用抑制剂的鉴定和验证及其体内抗转移作用
为了将机制研究成果转化为潜在的治疗策略,他们尝试通过计算机筛选方法寻找靶向ENO2-MIF相互作用的小分子抑制剂,构建了一个包含6723个化合物的化合物库,并依次通过吸收、分布、代谢、排泄和毒性(ADME/T)评估、多级分子对接以及基于广义Born和表面积溶剂化(MM-GBSA)的分子力学结合自由能重评分进行筛选(图8a)。在众多候选化合物中,吡硫氧二盐酸盐表现出最佳的预测结合亲和力之一(图8b)。在验证实验中,包括吡硫氧二盐酸盐在内的筛选化合物能够有效破坏ENO2与MIF在Co-IP实验中的相互作用,并增加MIF的泛素化水平(图8c)。分子对接表明吡硫氧嘧啶稳定地结合在 ENO2-MIF 界面(图8d),分子动力学模拟中稳定的均方根偏差 (RMSD) 和有利的残基特异性均方根波动 (RMSF) 特征进一步支持了这一预测(图8e)。
接下来,他们使用肝转移模型(图8f)评估了吡硫辛在体内的抗转移疗效。与溶剂对照组相比,口服吡硫辛(10 或 20 mg/kg)显著抑制了肝转移灶的形成,表现为生物发光信号减弱和肝表面转移结节数量减少(图8g)。组织学分析证实肝实质内的转移负荷显著降低。与 ENO2-MIF 轴在激活 STAT3 和 NF-κB 信号通路中的作用一致,吡硫辛治疗小鼠的肿瘤组织显示 MIF 水平降低,STAT3 和 P65 的磷酸化水平也降低(图8h)。这些结果表明吡硫辛是一种新型的 ENO2-MIF 相互作用抑制剂,并证实了其在体内具有强大的抗转移活性,凸显了靶向该蛋白-蛋白相互作用界面的治疗潜力。
总之,本研究揭示了CRLM的一种新机制:ENO2与MIF结合,抑制其CHIP介导的泛素化降解,从而激活MIF通路,诱导M2型巨噬细胞极化,进而驱动免疫抑制和转移性增殖。这种肝转移机制依赖于ENO2阳性癌细胞和M2型巨噬细胞的共同参与,而一种新型的ENO2-MIF相互作用抑制剂——吡硫氧嘧啶,能够有效干预肝转移过程。本研究确定ENO2是阻断CRLM的关键靶点。
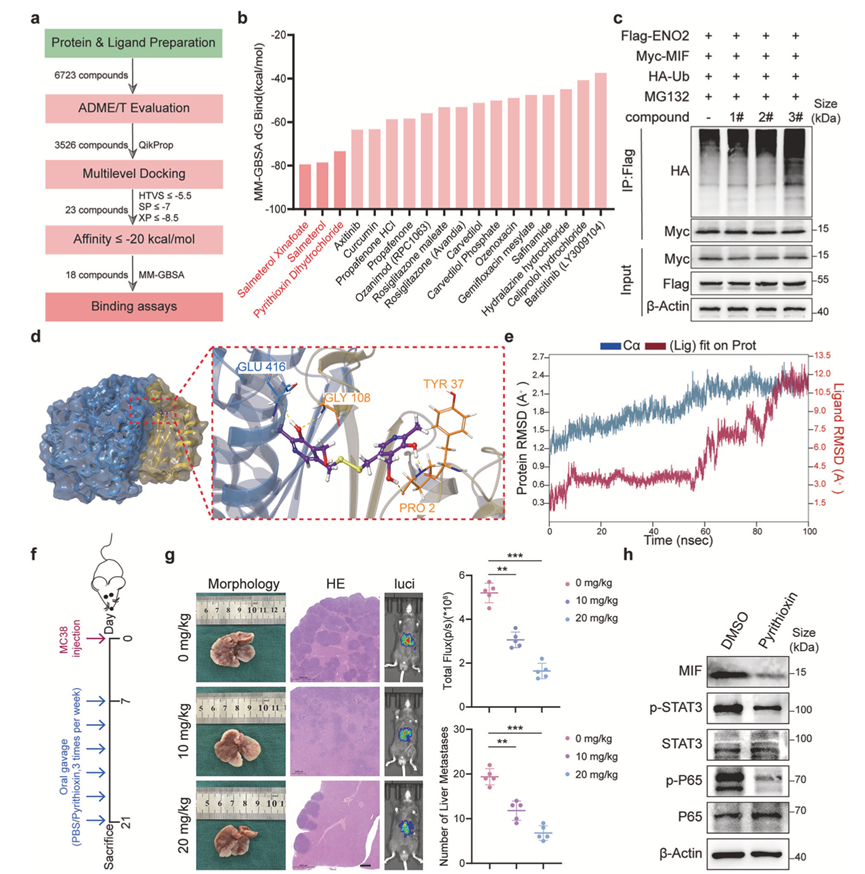
图8. 吡硫辛作为ENO2-MIF相互作用抑制剂的鉴定和验证及其体内抗转移作用。
(a) 计算机筛选策略的工作流程。共对6723个化合物进行ADME/T评价、多级分子对接、MM-GBSA重评分和结合实验,以筛选出高亲和力的候选化合物。(b) 排名靠前化合物的MM-GBSA结合自由能(ΔG_bind)值。(c) 免疫共沉淀( Co-IP)实验表明化合物(#1、#2和#3)破坏了ENO2和MIF之间的相互作用。(d) 分子对接模型展示了吡硫辛在ENO2-MIF界面内的结合模式,并突出显示了关键的相互作用残基。(e) ENO2-吡硫氧嘧啶复合物的分子动力学模拟显示了RMSD随时间的变化以及残基RMSF分析,表明该复合物具有稳定的结合。(f) 体内实验设计示意图。(g) 肝转移灶的代表性图像。(h) Western blot 分析。
+ + + + + + + + + + +
结 论
与非转移性CRC患者相比,CRLM患者中表达ENO2的癌细胞显著富集。生物信息学和小鼠模型支持的功能表征表明,ENO2+癌细胞表现出增强的上皮-间质转化,并且是CRLM的关键驱动因素。从机制上讲,ENO2蛋白直接与癌细胞内的MIF结合,通过抑制MIF C端Hsc70相互作用蛋白(CHIP)介导的泛素化和降解来稳定MIF。这种ENO2-MIF相互作用激活MIF信号通路,促进肿瘤细胞与巨噬细胞之间强烈的相互作用,进而促进M2型巨噬细胞极化。空间转录组学分析证实了ENO2阳性癌细胞与M2型巨噬细胞的共定位。至关重要的是,类器官模型和体内模型均证实,CRC细胞中的ENO2对于通过MIF通路诱导M2型巨噬细胞极化至关重要,从而促进肝转移。在小鼠模型中,ENO2基因敲除显著抑制了肿瘤生长和肝转移。ENO2-MIF相互作用抑制剂吡硫氧嘧啶(pyrithioxin)能够有效减轻小鼠肝转移的负担。总的来说,ENO2 通过稳定 MIF 来协调 M2 巨噬细胞极化,从而成为 CRLM 的关键驱动因素,凸显了 ENO2-MIF 轴作为 CRLM 的一种有前景的治疗策略。
+ + + + +
 ✦ +
✦ ++
论文ID
原名:ENO2 drives tumor cell-induced M2 macrophage polarization to promote colorectal cancer liver metastasis
译名:ENO2驱动肿瘤细胞诱导的M2巨噬细胞极化,从而促进结直肠癌肝转移
期刊:Signal Transduction and Targeted Therapy
影响因子:52.7
发表时间:2026.05.05
DOI号:10.1038/s41392-026-02732-2
背 景
结直肠癌(CRC)是全球第三大常见恶性肿瘤,每年新增病例超过190万例,死亡主要由远处转移引起。在转移部位中,肝脏是最常见且最致命的,占CRC相关死亡病例的70%以上。目前的一线全身治疗,包括抗表皮生长因子受体(EGFR)和抗血管内皮生长因子(VEGF)靶向药物,对结直肠癌肝转移(CRLM)患者的生存获益有限,尤其是对于绝大多数微卫星稳定型(MSS)CRC患者,这类患者对单药免疫检查点阻断疗法存在原发性耐药。虽然在手术切除、全身化疗和新兴免疫疗法等多模式治疗方面取得了进展,但CRLM的5年生存率仍然低于20%。这种严峻的预后凸显了迫切需要破译肝脏趋向性和免疫逃逸的分子驱动因素,这是实现转移性生长的关键生物学特征。
实验设计
结 果
01
单细胞图谱揭示肝转移病灶恶性程度增强
为了阐明结CRLM的细胞生态系统,研究团队对来自6例患者的15份组织标本进行了单细胞RNA测序(scRNA-seq):其中3例为非转移性结直肠癌(CRC-nM),3例为CRLM。对于CRLM患者,配对样本包括原发性结直肠肿瘤(CRC)、邻近肠道组织和肝转移灶(LM)。
在三例CRLM患者中,无监督聚类分析显示,上皮细胞是原发灶和转移灶肿瘤组织中的主要细胞成分(图1b-d)。研究团队进一步对上皮细胞进行亚聚类分析,发现肿瘤组织中癌细胞的富集程度高于邻近组织(图1e-f)。值得注意的是,与原发灶癌细胞相比,肝转移灶癌细胞表现出多方面的恶性增强证据。基因组不稳定性升高表现为拷贝数变异(CNV)评分显著升高(图1g),同时伴有增殖驱动基因和缺氧反应基因的转录上调(图1h)。这些分子改变最终导致功能增强的表型,转移灶细胞表现出显著增强的增殖能力和缺氧适应能力(图1i-j)。总的来说,单细胞图谱描绘了CRLM的细胞生态系统,并揭示了肝转移病灶的恶性程度增加,与原发肿瘤相比,其特征是基因组不稳定性更高、增殖能力更强、缺氧适应性更强。
图1. 单细胞图谱揭示肝转移灶恶性程度增强。
(a) 研究设计:对3例非转移性结直肠癌(CRC)患者和3例结直肠癌肝转移(CRLM)患者的组织进行单细胞RNA测序(scRNA-seq)。(b) 3例CRLM患者肝转移灶(LM)、邻近组织(CRC邻近组织)和原发性CRC组织中主要细胞类型的组成。(c) 11种细胞类型的UMAP可视化图,这些细胞类型由标记基因注释。(d) 注释细胞的组织来源分布。(e) 上皮细胞亚群分析显示肿瘤中癌细胞富集。(f) 上皮细胞亚群的比例丰度。(g) 原发性(CRC)和转移性(LM)部位癌细胞的拷贝数变异(CNV)评分。(h) 火山图显示scRNA-seq数据中差异表达的基因,比较肝转移灶(LM)细胞和原发性CRC细胞。(i) 转移性癌细胞与原发性癌细胞的细胞增殖特征。(j) 肝转移中缺氧相关基因表达。
02
基于模块的转移启动型癌细胞亚群发现
为了揭示转移启动的癌细胞亚群,他们对scRNA-seq数据进行了基于模块的分析。利用均匀流形逼近和投影(UMAP)方法对肿瘤细胞进行降维,结果显示转移灶和原发灶中均存在八个不同的细胞亚群(图2a-b),这些亚群表现出不同程度的恶性程度。
接下来,他们应用模块分析模型对癌细胞进行聚类,并将结果与上述转移相关亚组进行比较。在“模块11”中得分阳性的细胞主要富集于癌细胞亚组1、2和3中,这些亚组共同表现出转移性癌细胞的特征(图2d)。重要的是,根据多因素Cox回归模型(图2e),“模块11”中的几个基因与癌症基因组图谱(TCGA)结肠腺癌(COAD)数据集中的CRC患者预后不良显著相关。
他们进一步分析了转移性和非转移性患者原位病灶的单细胞谱。聚焦于原发病灶,UMAP降维分析识别出六个亚组,其分布在转移性和非转移性患者之间存在差异(图2f-g)。对这些亚组的经典恶性特征进行评估发现,癌细胞亚组3表现出显著的上皮-间质转化(EMT)特征,提示其可能在启动转移中发挥作用(图2h-i)。综上所述,这些结果表明“模块11”基因集符合转移性CRC细胞的表达特征,并明确指出具有EMT特征的独特癌细胞亚群(亚组3),该亚群可能代表原发肿瘤中一种转移启动状态。
图2. 基于模块的转移启动型癌细胞亚群发现。
(a-b) 对肿瘤细胞进行UMAP降维分析,并展示了转移灶和原发灶中八个机械亚群的分布。(c) 通过CNV评分评估八个癌细胞亚群的恶性程度,以突变负荷较低的巨噬细胞(MP)作为阴性对照。(d) 基于模块分析模型对癌细胞进行聚类和降维。(e) 使用TCGA-COAD数据集中的多因素Cox回归模型分析“模块11”基因集中的基因与结直肠癌预后的相关性。(f-g) 对来自 6 个原发病灶组织的肿瘤细胞进行了UMAP 降维分析。(h-i) 利用肿瘤细胞的典型恶性特征分析了六个肿瘤细胞亚群的潜在特征。
03
ENO2驱动转移进展,并可作为治疗靶点
基于转移相关特征的鉴定,他们从“模块11”和癌细胞3的表达谱中筛选出10个候选基因(图3a)。其中,只有烯醇化酶2(ENO2)证实为重要的预后决定因素,其高表达与CRC患者生存期显著缩短相关(图3b)。ENO2是一种糖酵解酶,催化2-磷酸甘油酸转化为磷酸烯醇式丙酮酸,正是这种双重性的一个例证。除了其在三磷酸腺苷 (ATP) 生成中的经典作用外,ENO2 在神经内分泌肿瘤、肺癌和CRC中均存在异常表达,并且与化疗耐药性相关。从机制上讲,ENO2 可通过核转位和组蛋白去乙酰化酶 (HDAC) 抑制来调节基因转录。ENO2 还可通过蛋白质-蛋白质相互作用稳定致癌激酶。然而,其作为CRLM中免疫微环境相互作用的关键调节因子的潜力尚未得到充分研究。南京和广州队列的临床验证证实,与正常组织相比,原发肿瘤和肝转移灶中ENO2表达均上调(图3c),并证实其与患者生存期呈负相关(图3d)。使用GSE41258数据集和TNMplot在线工具验证了ENO2表达与CRLM之间的相关性。在C57BL/6J小鼠模型中进行的功能性研究揭示了ENO2敲除对肿瘤转移的显著抑制作用。皮下异种移植瘤实验表明,ENO2敲除组的肿瘤生长显著减弱,表现为肿瘤体积和重量的降低(图3e)。更重要的是,脾脏注射模型显示,ENO2缺陷细胞的肝脏转移受到抑制,转移结节减少了50%以上(图3f)。
药理学抑制ENO2(ENOblock)进一步强调了其治疗意义,该抑制剂能有效阻断ENO2的促转移功能(图3g)。多重免疫荧光证实了ENO2与EMT诱导之间的机制联系,显示ENO2表达肿瘤中E-钙黏蛋白丢失和波形蛋白增加(图3h)。这表明靶向ENO2具有一定的临床应用前景。
图3. ENO2驱动转移进展并可作为治疗靶点。
(a) 从“模块11”基因集和“癌细胞3”特征基因中筛选出10个候选基因。(b) 基于TCGA-COAD数据库,采用多因素Cox回归分析这10个候选基因与结直肠癌预后的相关性。(c) 在多个独立队列中,采用免疫组化方法检测ENO2表达与结直肠癌进展的相关性。(d) 利用来自南京和广州的三个独立样本队列,进行Kaplan- Meier生存分析,探讨ENO2表达与结直肠癌预后的关系。(e) 在C57BL/6J小鼠皮下肿瘤模型中观察ENO2敲除DLD-1细胞的致瘤能力,并比较肿瘤组织的重量和体积。(f) 利用C57BL/6J小鼠脾内注射模型,观察了ENO2敲除DLD-1细胞与野生型细胞形成的肝转移结节数量。(g) 使用ENO2药理学抑制剂(ENOblock)验证靶向ENO2抑制结直肠癌肝转移(CRLM)的潜力。(h) 对对照组和ENO2调控的癌细胞进行EMT标志物(E-钙黏蛋白、波形蛋白)的多重免疫荧光染色。
04
ENO2 通过与 MIF 的直接相互作用来调控 M2 巨噬细胞极化
为了研究癌细胞如何塑造肿瘤微环境(TME),他们利用CellChat分析了细胞间通讯。分析结果显示,ENO2+癌细胞是TME中的主要信号枢纽,其中巨噬细胞迁移抑制因子(MIF)信号通路表现出特别强且特异性的外向/内向信号(图4a)。聚焦于MIF介导的相互作用,ENO2+癌细胞与巨噬细胞的通讯显著增强,而ENO2-癌细胞则不然(图4b-c)。这一计算预测结果与观察结果相符,即转移灶中的巨噬细胞比原发性或非转移性肿瘤中的巨噬细胞表现出更高的M2极化倾向(图4d)。
为了阐明其机制基础,他们通过共免疫沉淀(Co-IP)结合质谱分析鉴定了ENO2和MIF之间的直接物理结合(图4e-f)。体外谷胱甘肽S-转移酶(GST)下拉实验(使用纯化的重组蛋白)证实了这种相互作用是直接的(图4g)。为了确定分子界面,他们进行了分子对接模拟,预测了ENO2和MIF之间的特异性结合模式(图4h)。随后,他们使用两种蛋白的结构域特异性突变体进行了Co-IP实验,验证了这些预测,结果表明破坏预测的界面完全消除了它们的结合(图4i)。这些结果证实了癌细胞中 ENO2 和 MIF 之间存在直接的物理相互作用,并表明这种相互作用是 ENO2 +癌细胞向巨噬细胞增强 MIF 介导信号传导的基础,从而促进了 M2 极化的免疫抑制微环境。
图4. ENO2通过与MIF的直接相互作用调控M2巨噬细胞极化。
(a) 基于CellChat分析不同细胞类型参与微环境信号通讯的强度和潜在方式。(b-c) 在MIF信号通路参与的情况下,分析了ENO2-/-癌细胞与不同细胞类型之间的相互作用强度。(d) 与原位或非转移性原位病变相比,转移性病变和转移性原位病变中的巨噬细胞更倾向于M2极化。(e) 免疫共沉淀(Co-IP)检测证实了内源性ENO2与MIF之间的相互作用。(f) 对DLD-1细胞中内源性MIF和ENO2进行共免疫沉淀分析。(g) GST下拉实验表明MIF和ENO2在体外存在直接相互作用。(h-i) 分子对接模拟预测了ENO2和MIF之间的关键结合界面,并使用结构域特异性突变体进行相互免疫沉淀分析。
05
ENO2通过抑制泛素介导的降解来稳定MIF,从而激活促转移信号通路
接下来,他们研究了ENO2-MIF相互作用的功能后果。邻位连接分析(PLA)证实了癌细胞内源性ENO2-MIF相互作用的存在(图5a)。功能上,在DLD-1细胞中敲低ENO2导致MIF蛋白水平显著降低,而在HCT-116细胞中过表达ENO2则增加了MIF的丰度,表明ENO2正向调控MIF的稳态水平(图5b)。
鉴于已知MIF受泛素介导的蛋白酶体降解调控,他们探究了ENO2是否通过干扰该过程来稳定MIF。使用蛋白酶体抑制剂MG132处理后,ENO2敲低细胞中的MIF水平得以恢复,而ENO2过表达细胞中的MIF水平则进一步升高(图5c)。在MG132处理的细胞中,ENO2敲低显著增强了MIF的多聚泛素化,而ENO2过表达则产生了相反的效果(图5d)。
结构域定位实验证实,这种稳定性需要完整的结合界面,因为ENO2催化结构域突变体无法保护MIF免受泛素化修饰(图5e-f)。最后,他们研究了其对下游致癌信号通路的影响。ENO2表达的调控相应地改变了STAT3和NF-κB亚基P65的磷酸化水平(图5g),这两个关键通路均由MIF激活。这些数据表明,ENO2通过拮抗CHIP介导的多聚泛素化及其后续的蛋白酶体降解来结合并稳定MIF蛋白,从而增强癌细胞中MIF依赖的STAT3和NF-κB信号通路的激活。
图5. ENO2 通过抑制泛素介导的降解来稳定 MIF。
(a) 邻位连接分析 (PLA) 显示细胞内源性 ENO2 与 MIF 之间的相互作用。(b-g) Western blot 分析。
06
ENO2诱导M2巨噬细胞极化,从而促进肝转移
为了探究ENO2-MIF轴是否在功能上重塑肿瘤免疫微环境,他们首先分析了二者的空间关系。空间转录组分析(ST)显示,在转移灶中,ENO2阳性癌细胞与M2样巨噬细胞存在显著的共定位(图6a)。对多个ST样本进行定量相关性分析证实,这两个细胞群之间存在一致且显著的正相关性。这种空间关联促使他们进一步探究表达ENO2的癌细胞是否能够直接诱导M2极化。
利用体外共培养系统,他们发现与ENO2敲低癌细胞共培养的巨噬细胞表现出M1标志物(CD86)增加和M2标志物(CD206)减少,而与ENO2过表达细胞共培养的巨噬细胞则呈现相反的趋势(图6b)。这种ENO2依赖的M2极化在体内也得到了重现,表现为与对照组相比,ENO2过表达细胞来源的异种移植瘤中CD206+巨噬细胞的比例更高(图6c)。使用患者来源的类器官(PDO)进一步证实了癌细胞内源性ENO2的关键作用;与 ENO2 敲除 PDO 共培养的巨噬细胞的 M2 表型降低,而与 ENO2 过表达 PDO 共培养的巨噬细胞则表现出增强的 M2 极化(图6d)。
随后,他们评估了该轴对体内转移的功能性影响。在C57BL/6J小鼠脾移植-肝转移模型中,癌细胞中ENO2的过表达显著促进了肝转移的形成(图6e-f)。重要的是,这种促转移作用可由MIF抑制剂ISO-1或M2极化抑制剂LPS消除。相反,ENO2敲低导致的转移减弱可通过给予M2激动剂IL-4或在癌细胞中强制过表达MIF来恢复(图6e-f),免疫组织化学证实了各组间M2极化的丰富程度。这些结果表明,ENO2阳性癌细胞通过分泌MIF驱动肿瘤微环境中M2巨噬细胞的极化,进而促进肝转移的形成。
图6. ENO2诱导M2巨噬细胞极化驱动肝转移。
(a) 空间特征图显示了ST_liver4中ENO2+癌细胞和M2巨噬细胞的反卷积比例。(b-c) 流式细胞分析共培养系统和异种移植瘤中巨噬细胞标志物CD86和CD206的表达。(d) 代表性流式细胞图显示了与患者来源类器官(PDO)模型共培养的巨噬细胞上CD206和CD86的表达。(e-f) 利用C57BL/6J小鼠脾脏移植模型,研究ENO2表达对肝转移形成的影响。
07
NO2 募集 HSP90 以拮抗 CHIP 介导的 MIF 泛素化和降解
为了进一步阐明ENO2如何稳定MIF,他们试图鉴定其分子互作蛋白。质谱分析ENO2复合物发现分子伴侣热休克蛋白90 (HSP90) 是其结合蛋白(图7a),这一结果通过内源性蛋白的双向免疫共沉淀(Co-IP)实验得到证实(图7b-c)。有趣的是,Co-IP实验表明ENO2增强了HSP90与MIF之间的物理结合,提示ENO2可能作为支架蛋白募集HSP90至MIF(图7d)。
由于已知E3连接酶CHIP靶向HSP90底物蛋白进行降解,他们假设ENO2可能通过拮抗CHIP来保护MIF。与此一致,ENO2敲低导致的MIF水平降低可通过同时敲低CHIP而恢复(图7e),而ENO2过表达诱导的MIF积累则可通过共表达CHIP而抑制(图7f)。从机制上讲,ENO2敲低显著增强了CHIP介导的MIF多聚泛素化(图7g)。
为了确定泛素化位点,他们构建了MIF突变体,其中候选赖氨酸残基(K32、K66、K77)被替换。泛素化实验表明,K66位点的突变(而非K32或K77位点的突变)显著抑制了CHIP介导的MIF多聚泛素化(图7h-i),从而确定K66为主要泛素化位点。综上所述,这些结果表明ENO2募集HSP90至MIF,形成三元复合物,该复合物竞争性抑制E3泛素连接酶CHIP,从而抑制CHIP介导的MIF在K66位点的泛素化及其后续的蛋白酶体降解。
图7. ENO2 募集 HSP90 以拮抗 CHIP 介导的 MIF 泛素化和降解。
(a) 代表性的液相色谱-串联质谱 (LC-MS/MS) 图谱,鉴定出 HSP90 为 ENO2 相互作用蛋白,并突出显示了匹配的肽段片段。(b-i) 免疫沉淀和Western blot 分析。
05
吡硫辛作为ENO2-MIF相互作用抑制剂的鉴定和验证及其体内抗转移作用
为了将机制研究成果转化为潜在的治疗策略,他们尝试通过计算机筛选方法寻找靶向ENO2-MIF相互作用的小分子抑制剂,构建了一个包含6723个化合物的化合物库,并依次通过吸收、分布、代谢、排泄和毒性(ADME/T)评估、多级分子对接以及基于广义Born和表面积溶剂化(MM-GBSA)的分子力学结合自由能重评分进行筛选(图8a)。在众多候选化合物中,吡硫氧二盐酸盐表现出最佳的预测结合亲和力之一(图8b)。在验证实验中,包括吡硫氧二盐酸盐在内的筛选化合物能够有效破坏ENO2与MIF在Co-IP实验中的相互作用,并增加MIF的泛素化水平(图8c)。分子对接表明吡硫氧嘧啶稳定地结合在 ENO2-MIF 界面(图8d),分子动力学模拟中稳定的均方根偏差 (RMSD) 和有利的残基特异性均方根波动 (RMSF) 特征进一步支持了这一预测(图8e)。
接下来,他们使用肝转移模型(图8f)评估了吡硫辛在体内的抗转移疗效。与溶剂对照组相比,口服吡硫辛(10 或 20 mg/kg)显著抑制了肝转移灶的形成,表现为生物发光信号减弱和肝表面转移结节数量减少(图8g)。组织学分析证实肝实质内的转移负荷显著降低。与 ENO2-MIF 轴在激活 STAT3 和 NF-κB 信号通路中的作用一致,吡硫辛治疗小鼠的肿瘤组织显示 MIF 水平降低,STAT3 和 P65 的磷酸化水平也降低(图8h)。这些结果表明吡硫辛是一种新型的 ENO2-MIF 相互作用抑制剂,并证实了其在体内具有强大的抗转移活性,凸显了靶向该蛋白-蛋白相互作用界面的治疗潜力。
总之,本研究揭示了CRLM的一种新机制:ENO2与MIF结合,抑制其CHIP介导的泛素化降解,从而激活MIF通路,诱导M2型巨噬细胞极化,进而驱动免疫抑制和转移性增殖。这种肝转移机制依赖于ENO2阳性癌细胞和M2型巨噬细胞的共同参与,而一种新型的ENO2-MIF相互作用抑制剂——吡硫氧嘧啶,能够有效干预肝转移过程。本研究确定ENO2是阻断CRLM的关键靶点。
图8. 吡硫辛作为ENO2-MIF相互作用抑制剂的鉴定和验证及其体内抗转移作用。
(a) 计算机筛选策略的工作流程。共对6723个化合物进行ADME/T评价、多级分子对接、MM-GBSA重评分和结合实验,以筛选出高亲和力的候选化合物。(b) 排名靠前化合物的MM-GBSA结合自由能(ΔG_bind)值。(c) 免疫共沉淀( Co-IP)实验表明化合物(#1、#2和#3)破坏了ENO2和MIF之间的相互作用。(d) 分子对接模型展示了吡硫辛在ENO2-MIF界面内的结合模式,并突出显示了关键的相互作用残基。(e) ENO2-吡硫氧嘧啶复合物的分子动力学模拟显示了RMSD随时间的变化以及残基RMSF分析,表明该复合物具有稳定的结合。(f) 体内实验设计示意图。(g) 肝转移灶的代表性图像。(h) Western blot 分析。
+ + + + + + + + + + +
结 论
与非转移性CRC患者相比,CRLM患者中表达ENO2的癌细胞显著富集。生物信息学和小鼠模型支持的功能表征表明,ENO2+癌细胞表现出增强的上皮-间质转化,并且是CRLM的关键驱动因素。从机制上讲,ENO2蛋白直接与癌细胞内的MIF结合,通过抑制MIF C端Hsc70相互作用蛋白(CHIP)介导的泛素化和降解来稳定MIF。这种ENO2-MIF相互作用激活MIF信号通路,促进肿瘤细胞与巨噬细胞之间强烈的相互作用,进而促进M2型巨噬细胞极化。空间转录组学分析证实了ENO2阳性癌细胞与M2型巨噬细胞的共定位。至关重要的是,类器官模型和体内模型均证实,CRC细胞中的ENO2对于通过MIF通路诱导M2型巨噬细胞极化至关重要,从而促进肝转移。在小鼠模型中,ENO2基因敲除显著抑制了肿瘤生长和肝转移。ENO2-MIF相互作用抑制剂吡硫氧嘧啶(pyrithioxin)能够有效减轻小鼠肝转移的负担。总的来说,ENO2 通过稳定 MIF 来协调 M2 巨噬细胞极化,从而成为 CRLM 的关键驱动因素,凸显了 ENO2-MIF 轴作为 CRLM 的一种有前景的治疗策略。
+ + + + +


