

 English
English文献解读|Cancer Cell(44.5):缺氧影响转移性透明细胞肾细胞癌的治疗反应和耐药性
✦ +
+
论文ID
原名:Hypoxia shapes both therapeutic response and resistance in metastatic clear cell renal cell carcinoma
译名:缺氧影响转移性透明细胞肾细胞癌的治疗反应和耐药性
期刊:Cancer Cell
影响因子:44.5
发表时间:2026.06.04
DOI号:10.1016/j.ccell.2026.05.007
背 景
血管内皮生长因子受体酪氨酸激酶抑制剂(VEGFR-TKI)和免疫检查点抑制剂(ICI)联合用药(简称TKI/IO)是多种实体瘤的标准治疗方案,包括肾细胞癌、子宫内膜癌和肝细胞癌。然而,对于许多此类癌症,TKI /IO联合用药的持久缓解率低于ICI单药治疗,且许多III期临床试验未能将II期临床试验中观察到的早期疗效转化为长期获益。在一项转移性黑色素瘤的III期研究中, TKI /IO联合用药组的疾病进展死亡率高于帕博利珠单抗(抗程序性死亡受体1;aPD-1)单药治疗组,这凸显了迫切需要深入了解联合用药和单药治疗的机制基础。它们的作用是叠加还是协同,以及它们如何随时间推移调节肿瘤微环境(TME)并影响长期预后的具体机制尚不清楚。这些见解对于开发合理的生物标志物和改进未来的治疗策略至关重要。
实验设计
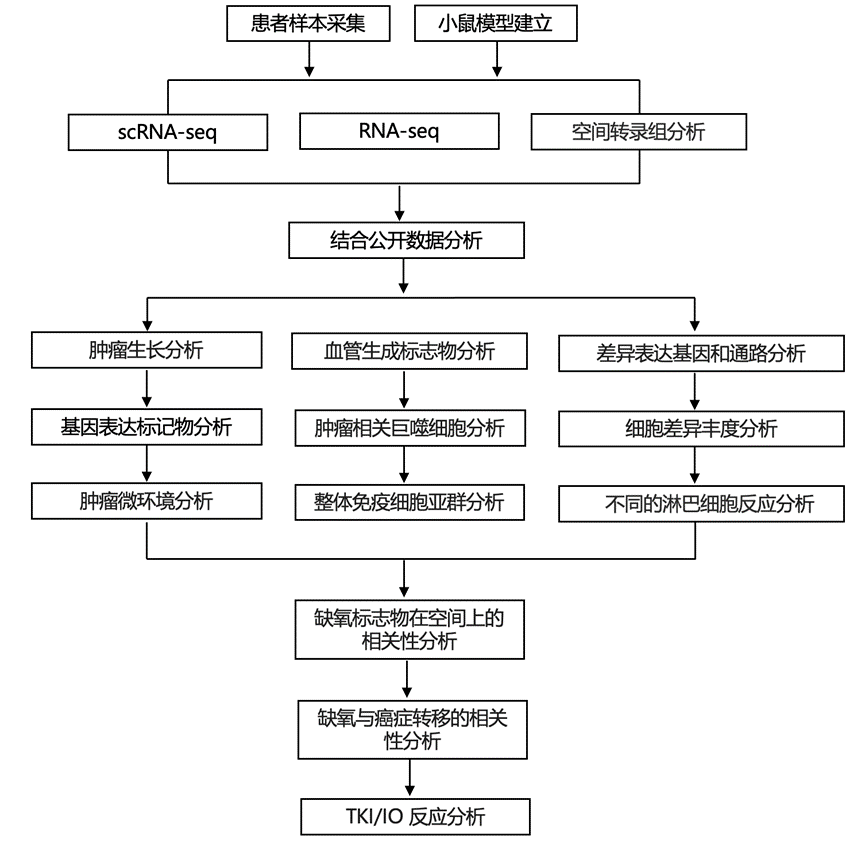
结 果
01

小鼠KVpR模型类似于人类难治性透明细胞肾细胞癌亚型
研究团队使用了Ksp-CreERT2;Vhlf/f;Trp53f/f;Rb1f/f (KVpR)小鼠,这些小鼠在5-12周内会发展出多个高级别肾脏肿瘤,其组织学和分子特征与人类透明细胞肾细胞癌(ccRCC)高度相似。为了表征该模型中的治疗反应,他们将荷瘤小鼠随机分为三组,分别接受标准一线TKI/IO联合治疗方案VEGFR-TKI乐伐替尼(Len)+抗PD-1抗体(aPD-1、单药治疗或不治疗(未治疗组,UT)。他们使用磁共振成像(MRI)对来自68只小鼠的共220个肿瘤进行了长达225天的纵向追踪。基于肿瘤生长阈值建立了小鼠肿瘤反应分类标准,并在急性或慢性治疗时间点收集部分肿瘤进行单细胞转录组分析 (scRNA-seq)(图 1A)。总共分析了来自 35 个肿瘤和2个正常肾脏 (NK)的231993 个细胞,他们还进行了免疫荧光染色 (IF) 和进一步的功能验证(图 1A)。同时,他们收集了13例接受一线TKI/IO或IO/IO治疗的患者在治疗期间的肿瘤样本,这些患者在延长治疗后接受了巩固性细胞减灭性肾切除术(图1A)。手术时,对样本进行了scRNA-seq分析,并在部分病例中进行了Visium空间转录组学(ST)分析(图1A)。为了进行比较,我们纳入了先前报道的两例未经治疗的肿瘤样本。他们使用另一个包含50例接受IO/IO或TKI/IO治疗的患者的治疗期间肿瘤样本的成像质谱流式细胞(IMC)队列验证了基于RNA的研究结果(图1A)。对于生物标志物研究,我们收集了已发表的临床试验中治疗前 ccRCC 肿瘤的转录组分析(RNA-seq)数据,以及来自未发表的 MSKCC 队列的 44 个治疗前肿瘤和 17 个治疗中肿瘤(图 1 A)。
鉴于ccRCC的分子异质性,他们首先利用RNA-seq技术检测了先前已鉴定的分子亚组中相关基因的表达情况。与NK细胞相比,KVpR肿瘤中T效应细胞和血管生成相关基因的表达较低,但细胞周期和髓系炎症相关基因的表达显著上调(图S1A)。根据基质基因的有无,可区分出两个亚聚类,这表明KVpR肿瘤与人类的“基质/增殖型”和“增殖型”亚聚类最为相似。这两个亚型约占ccRCC的20%,TP53突变的富集程度相似,并且通常对某些VEGF/IO联合疗法最不敏感。
流式细胞术证实了T细胞数量稀少和髓系细胞占优势(图S1B-C)。CD4+T细胞主要由Treg细胞和Th1细胞组成(图S1 D),而大多数CD8+ T细胞为组织驻留细胞(CD49a+),但缺乏活化(PD-1+)或细胞毒性(颗粒酶B +)细胞(图S1 E)。有趣的是,超过50%的成熟肿瘤相关巨噬细胞(TAM)缺乏MHC II类分子且PD-L1+(图S1F),表明其处于免疫抑制状态。他们进一步观察到PD-1及其配体PD-L1在髓系细胞和淋巴细胞群中广泛表达(图S1G)。因此,KVpR模型反映了一种侵袭性强、髓系细胞占优势、适应性免疫抑制的肿瘤微环境(TME)模型,为研究治疗耐药性ccRCC亚型提供了一种有价值的工具。
对KVpR小鼠肿瘤生长的追踪显示,对照组肿瘤呈指数增长,大多数肿瘤在约65天时达到终点(图1B)。与免疫抑制性TME一致,肿瘤对aPD-1治疗无显著反应,尽管存活至65天后的小鼠数量有所增加(图1B)。相反,所有肿瘤对Len均高度敏感,病情在第50天前保持稳定,但无一例外地在65天至220天以上出现耐药性。aPD-1/Len联合治疗虽不能治愈肿瘤,但显著延缓了耐药性的出现至200天以上(图1B),提示二者可能存在叠加或协同效应。从无应答者 (NR)、部分应答者 (PR) 或应答者 (R) 中收集肿瘤,用于后续的 IF 和 scRNA-seq 比较,同时与接受 2 周治疗的急性早期无应答者 (eNR) 或早期应答者 (eR) 进行比较(图1A,图S1H)。
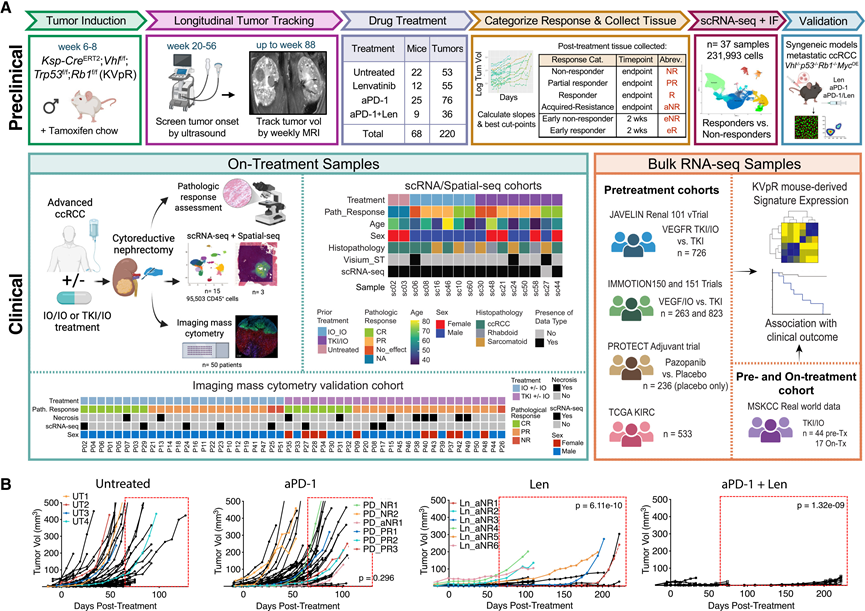
图1. KVpR 模型中的研究概述和治疗反应。
(A) 示意图。(B) KVpR 肿瘤生长曲线。
图S1. 分子聚类和肿瘤分析。
(A) 使用基因捕获的KVpR肿瘤和正常肾脏的RNA-seq技术比较不同的分子聚类。(B-G) 通过流式细胞技术对KVpR肿瘤浸润免疫细胞进行表征。(H)小鼠肿瘤体积生长曲线。
02
VEGFR-TKI 可促进小鼠肿瘤内严重缺氧
与直觉相反,血管生成靶向药物能够“正常化”结构渗漏和异常的肿瘤血管,从而改善灌注并减轻缺氧驱动的促肿瘤过程,后者可导致免疫抑制、上皮间质转化(EMT)和转移。然而,高临床剂量下是否会发生血管正常化(VN)尚存争议,且临床证据匮乏。此外,VN 的功能影响可能取决于癌症类型,而且不幸的是,定量ccRCC中真正的肿瘤缺氧尤其困难,因为许多其他癌症中常用的缺氧标志物位于 VHL 的下游。此外,鉴于假性缺氧 ccRCC 本身具有较高的血管生成能力,关于基线血管功能、VEGFR-TKI 治疗后引起的改变、由此导致的缺氧以及对短期和长期生存的影响等基本问题仍未得到充分探索。
为了解答这些悬而未决的问题,他们利用全身性注射匹莫尼达唑(也称缺氧探针或HPP)对KVpR肿瘤中的真正缺氧情况进行了可视化和定量分析。匹莫尼达唑在低氧条件下可与组织结合。HPP与血管标志物CD31和周细胞标志物NG2的共免疫荧光染色显示,令人惊讶的是,KVpR肿瘤在基线水平通常非常低(图2A-C)。与此同时,肿瘤内广泛存在血管生成(图2A-D),血管呈均匀分支状,并由周细胞(一种常用的血管生成标志物)提供结构支撑(图2A-E)。他们观察到局灶性缺氧区域,其缺氧程度随肿瘤体积呈指数级增长(图2B),但这与癌细胞增殖增加无关(图2F)。综上所述,这些结果表明,ccRCC的微血管网络功能强大,能够有效地支持肿瘤生长,直至肿瘤最终超出其血液供应能力。
与未治疗的肿瘤相比,长期使用抗PD-1治疗可降低VN,但并未显著改变缺氧或血管生成(图2A-E)。有趣的是,即使在较低剂量下,Len或抗PD-1/Len联合治疗也能有效破坏肿瘤血管(图2G,图S1H),导致CD31 +血管远端出现广泛的缺氧和坏死(图2A-D),但并未影响VN(图2E)。长期Len暴露维持了持续的血管修剪和缺氧,尽管在某些肿瘤中血管生成有所反弹,这表明癌细胞存在适应性选择(图2A-D)。停止治疗后,肿瘤血管恢复并逆转了缺氧,导致肿瘤快速再生(肿瘤Ln_aNR1,第166天)(图1B)。总之,ccRCC 肿瘤灌注效率很高,但 Len 却能有效破坏它,这与 TKI 的 VN 效应存在争议相反。
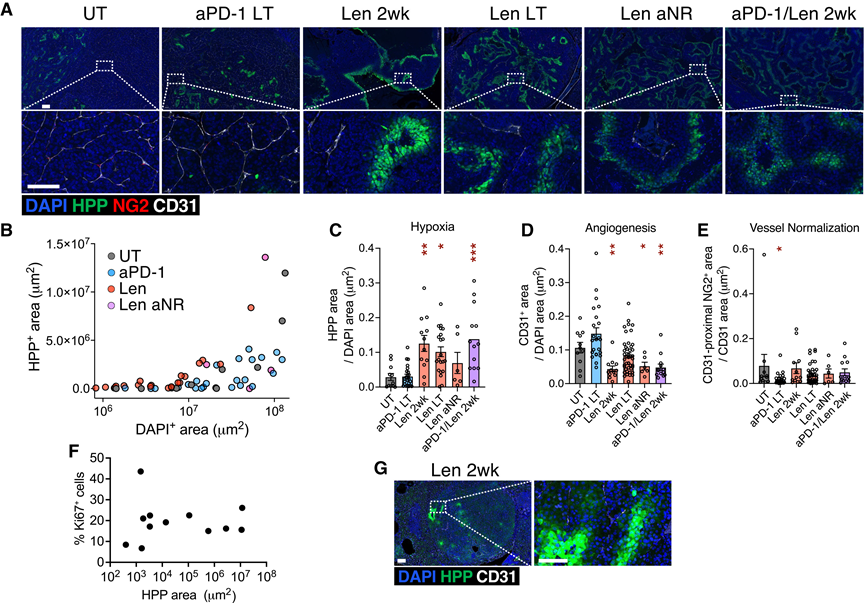
图2. 乐伐替尼促进严重的肿瘤内缺氧。
(A) KVpR 肿瘤的 IF 染色。(B) HPP 面积与 DAPI 面积的相关性。(C-E) 缺氧、血管生成和血管正常化的定量分析。(F) Ki67+细胞百分比与未治疗对照肿瘤的缺氧面积之间的关联。(G) 图示为3只KVpR小鼠肿瘤的代表性免疫荧光染色结果。
03
TKI 和 IO 对肿瘤免疫微环境的调节作用不同
为了初步了解治疗和缺氧相关的变化,他们利用scRNA-seq对KVpR小鼠和人ccRCC中的肿瘤浸润免疫细胞进行了正交分析。小鼠分析结果与他们的转录组和流式细胞数据一致(图S1),显示未经治疗的肿瘤中髓系细胞占优势,而NK细胞较少(图3A-C)。虽然缺氧诱导方式不同,但治疗后有反应的肿瘤表现出一些共同特征,与药物类别无关,包括中性粒细胞和T细胞数量增加,以及肿瘤相关巨噬细胞(TAM)数量减少(图3D)。他们还观察到一些药物特异性效应,例如抗PD-1治疗组(aPD-1)中B细胞数量增加(图3D)。Len特异性效应包括Len组和aPD-1/Len联合治疗组(aPD-1/Len)中浆细胞和cDC1细胞富集(图3D)。Len还能增加肝细胞癌中的树突状细胞(DC)数量,这与已知的缺氧对DC的免疫调节作用相符。对 aPD-1 或aPD-1/Len 有反应的40 名患者 CD4+和 CD8+ T 细胞较多,但有趣的是,单药 Len 治疗并未观察到这种情况(图 3 D)。
正如预期,他们观察到人类肿瘤的变异性更大,且T细胞数量显著高于小鼠(图3E-H)。总体而言,与未治疗的肿瘤相比,IO/IO和TKI/IO治疗均促进了Treg、CD8+ T细胞和B/浆细胞的浸润,但IO/IO治疗组的CD8 + T细胞和B/浆细胞反应明显优于TKI/IO治疗组(图3G),这与小鼠的结果基本一致(图3D)。TKI/IO治疗还具有其他特异性效应,包括CD4+ T细胞和自然杀伤(NK)细胞/固有淋巴细胞(ILC)数量的增加,而巨噬细胞则在IO/IO治疗后特异性富集(图3G)。值得注意的是,人类样本中中性粒细胞数量较少(图3C-G)。鉴于治疗期间采样时间各异且患者间存在异质性,很难辨别主要免疫细胞群与疗效之间是否存在显著且一致的关联(图 3H)。免疫抑制剂 (IO) 和酪氨酸激酶抑制剂 (TKI) 对免疫TME的影响可能存在差异,且具有叠加效应,这些效应在人和小鼠中大多重叠。
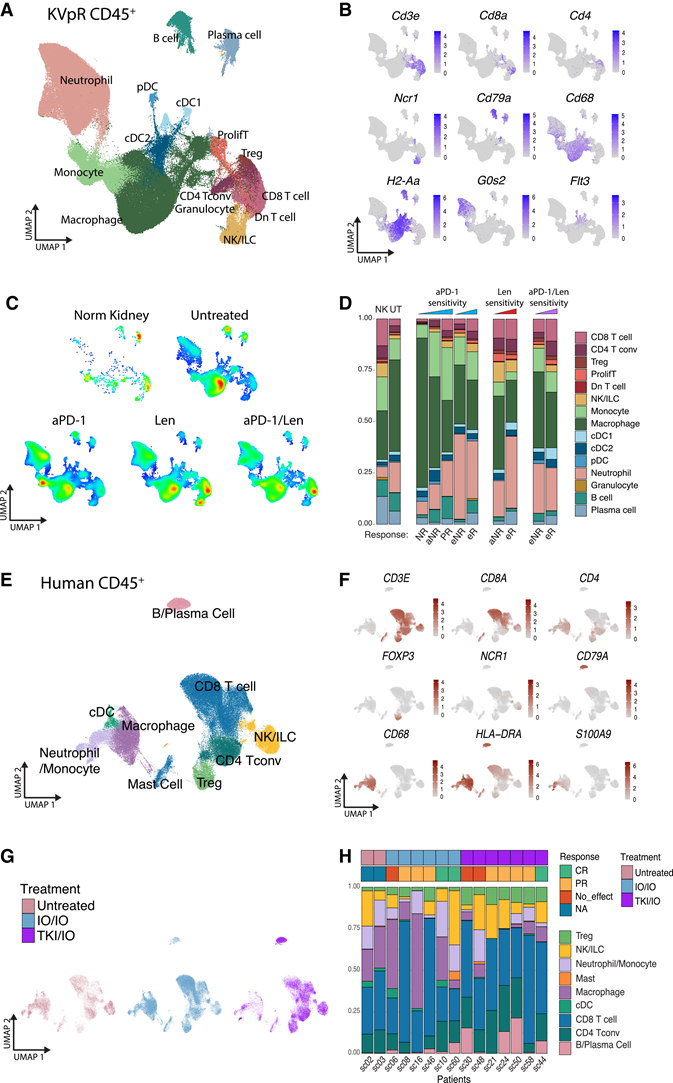
图3. TKI和IO对肿瘤免疫微环境的调节作用存在差异。
(A-H) 对KVpR小鼠或人透明细胞肾细胞癌(ccRCC)肿瘤中所有CD45+细胞进行scRNA-seq分析。
04
标题SPP1+ TAM在真正的缺氧条件下分化,并在TKI反应性小鼠肿瘤中增加
接下来,他们将分析重点放在TAM上,因为大量研究表明TAM与T细胞功能障碍、转移进展以及肾细胞癌(RCC)中免疫疗法(IO)或酪氨酸激酶抑制剂(TKI)疗法的耐药性有关。单核细胞、巨噬细胞和DC的聚类分析揭示了22个不同的细胞群,包括TAM、cDC1、cDC2、浆细胞样树突状细胞(pDC)和肥大细胞。尽管之前已去除双细胞群,但仍有一些双细胞群存在,这些双细胞群共表达TAM/DC标志物以及淋巴细胞谱系基因Cd3g和Nkg7(图4A-B)。对五个TAM亚聚类(不包括增殖型、双联型和中间型TAM,因为它们缺乏明显的差异表达基因)的功能分析显示,存在两个主要亚聚类:mM0,其特征是抗原呈递和适应性免疫应答通路,并表达Selenop、Mrc1 (CD206)和C1qa;以及mM1,其特征是血管生成、糖酵解和伤口愈合通路,并表达Spp1、Gpnmb和Arg1(图4A-C)。其他TAM亚聚类则表现出混合功能或干扰素应答功能(图4C)。
他们评估了不同治疗组和应答组中的细胞聚类比例(图 4D),并分别在 aPD-1 和 Len 组中使用 Milo 算法定量了不同应答组的差异丰度(图 4E)。正如在总 CD45+水平上观察到的(图 3D),他们在 Len 和 aPD-1/Len 应答者中观察到 mM13 和 mM14 cDC1 细胞聚类增加(图 4D-E)。出乎意料的是,他们观察到在所有治疗组中,抗原呈递细胞 mM0 Selenophi TAM在 NR 组中相对于应答者升高(图4D-E)。另一方面,mM1 Spp1+ TAM 在未治疗的肿瘤中水平较低,在 aPD-1 NR 组中富集,但在 Len 和 aPD-1/Len 应答者中显著富集(图 4E),这与 IF HPP 染色观察到的总体模式非常相似(图 2)。这促使他们假设 mM1 TAM 的募集是对真实微环境缺氧的响应,而非 VHL 缺失驱动的 VEGF 信号通路。事实上,在缺氧培养箱中培养原代人单核细胞来源的巨噬细胞 (hMDM) 可促进 SPP1 的分泌(图 4F),并显著增加 mM1 TAM 基因特征的表达,同时降低 mM0 基因特征的表达(图 4G)。化学缺氧模拟剂 CoCl2的作用较弱(图 4F),这些结果共同支持了 mM1 Spp1⁺ TAM 的分化是由低氧条件触发的,并且在功能上与 mM0 TAM 相反。相应地,在 KVpR 肿瘤中,血管生成性中性粒细胞也与 mM1 TAM 同步增加,并且 SPP1⁺ TAM与人类肿瘤中的缺氧相关,这进一步支持了这些细胞对真实组织缺氧具有特异性。因此,SPP1+ TAM 可能作为治疗反应性肿瘤中 Len 诱导的缺氧坏死的替代指标,以及 aPD-1 耐药和未治疗肿瘤中与肿瘤大小相关的缺氧坏死的替代指标。
在患者TAM中,mM0 和 mM1 特征呈中度负相关(图 4H),与 KVpR 模型中观察到的趋势一致。通过分析治疗反应与特征表达的关系,发现 TKI/IO 治疗可增加所有有效患者(部分缓解/完全缓解)TAM 中 mM1 特征的表达(图 4I),提示在人和小鼠中,与治疗疗效相关的缺氧巨噬细胞反应相似。
为了评估SPP1+ TAM与KVpR肿瘤中真正缺氧的空间共定位,他们对SPP1、CD68、CD31和HPP进行了共染色,发现癌细胞的SPP1标记强度高于CD68+巨噬细胞(图S2 H)。然而,GPNMB(mM1聚类中排名第二的差异表达基因)在从RCC肿瘤中分选出的细胞群中对巨噬细胞的特异性更高(图4 J)。虽然免疫荧光染色在小鼠中未成功,但人特异性GPNMB抗体对CSF1R+细胞具有特异性,并且在患者肿瘤中强烈标记了缺乏CD31+血管的坏死区域,类似于小鼠的HPP(图4 K)。此外,在与坏死无关的点状区域也观察到了GPNMB+ TAM。
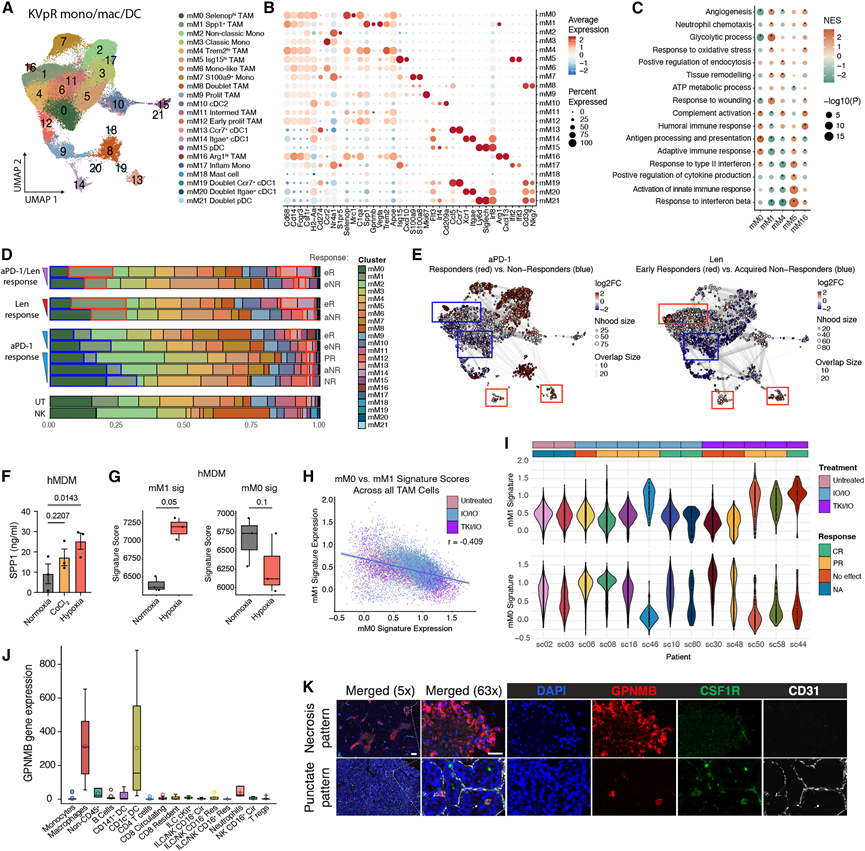
图4. SPP1+ GPNMB+ TAM与真正的缺氧和 TKI 治疗反应相关。
(A-B) 小鼠 KVpR 肿瘤中单核细胞、巨噬细胞和树突状细胞的 scRNA-seq UMAP和谱系定义和功能标记的散点图。(C) 点图描绘了主要巨噬细胞簇中生物通路的归一化富集分数。(D) 治疗反应组中髓系细胞簇的堆叠条形图。(E) 使用 Milo 计算的髓系细胞邻域的 UMAP 嵌入。(F-G) 用 ELISA 测量用 CoCl 2或在缺氧室中培养的 hMDM 的上清液或批量 RNA-seq中的 SPP1。(H) 人类 scRNA-seq 队列中所有 TAM 中小鼠 mM0与mM1特征表达的散点图和 Spearman 相关性。(I) 按患者绘制所有髓系细胞中 mM0 和 mM1 基因特征的小提琴图。(J) 通过流式分选的肾细胞癌肿瘤来源细胞群进行批量RNA-seq,检测GPNMB的表达。(K) 代表性的未经治疗的人类RCC肿瘤免疫荧光染色图。
05
小鼠治疗结果相关的淋巴细胞反应
鉴于缺氧对淋巴细胞功能的抑制作用,他们对KVpR肿瘤中的T细胞和固有淋巴细胞进行了亚群分析(图5A-B)。虽然不同反应组的总T细胞丰度存在差异(图3D),但各个T细胞和NK细胞亚群的比例基本保持不变(图5C)。与aPD-1处理的肿瘤相比,Len处理的肿瘤中CD8+ T细胞的活化和耗竭标志物表达升高(图5D),这与cDC1诱导增强相一致(图4A -E)。虽然假设NR中抗原呈递细胞Selenop hi TAM丰度的增加可能导致T细胞耗竭,但这一假设并不明显(图5D),提示存在T细胞非依赖性的抑制作用。
在小鼠(图 3D)和人类(图 3G)中,TKI 治疗后浆细胞/B 细胞数量增加。鉴于它们在识别细胞内抗原方面的作用,他们假设它们的募集反映了Len诱导的缺氧坏死后抗原的释放。支持这一假设的是,在治疗有效的肿瘤中,与抗 PD-1 抗体不同,Len不会诱导裂解型 caspase-3 介导的细胞凋亡,这是一种非免疫原性细胞死亡形式。对 Th1/Tfh 样 CD4+细胞聚类(mL2) 的分析显示,不同反应组之间细胞丰度或极化无差异,浆细胞分化的关键调节因子也无变化(图 5C)。无论是否接受治疗,应答者均表现出Tbx21 (Tbet) 表达增加,而只有 TKI/IO 诱导Pdcd1、Tnf和Ifng的同步上调,支持联合治疗特有的 Th1 偏向反应。
他们对所有淋巴细胞进行了亚群分析(图5E-F)。通过定量两两治疗组间的细胞群差异,他们发现,与TKI/IO组相比,IO/IO组样本中细胞毒性CD8+亚群增加,包括耗竭的hL0亚群、先前报道的ZNF683 +组织驻留记忆样hL2亚群、MKI67+增殖的hL9亚群以及表达干扰素刺激基因(ISG)的hL12亚群(图5G)。此外,虽然检测到的浆细胞数量较少,但B细胞亚群hL7和hL14以及CD4+ Tfh样hL10亚群在TKI/IO组和IO/IO组样本中的丰度均显著高于未治疗的对照组,且在TKI/IO组中更为显著(图5G)。最近一项研究表明,抗CTLA-4 反应与肿瘤引流淋巴结中 Treg 细胞的耗竭相关,这使得RCC中干细胞样 CD4+ T 细胞能够向 Th1 细胞分化。因此,他们应用 Palantir 轨迹检测算法分析了非 Treg CD4+细胞聚类,以研究治疗对 CD4+ T 细胞分化的影响。与所提出的模型一致,Palantir 推断出 Th1 和 Tfh 样谱系之间存在分化(图 5H),并且观察到在接受 IO/IO 治疗的患者中,Th1 轴早期阶段的分布发生了偏移,但末端阶段的分布没有变化(图 5I)。
在所有样本中,B细胞和Tfh样细胞的比例呈正相关,在TKI/IO治疗的患者中水平最高。对两个TKI/IO样本和一个IO/IO样本进行ST分析证实了B细胞和Tfh样细胞的空间共定位,且在TKI/IO样本中丰度更高。病理学家注释证实这些区域存在密集的淋巴细胞浸润,但没有生发中心,提示B细胞/Tfh细胞相互作用可能发生在TLS前结构或所谓的淋巴-髓系聚集体中。结合浆细胞的缺乏以及B细胞中抗原呈递机制的高表达但未发生类别转换的免疫球蛋白产生(图5 F),这提示存在早期或已消退的抗体反应。总之,他们的人体数据与小鼠研究结果基本一致,即IO主要诱导CD8+ T细胞活化,而TKI促进B细胞活化,这可能反映了缺氧诱导的坏死。
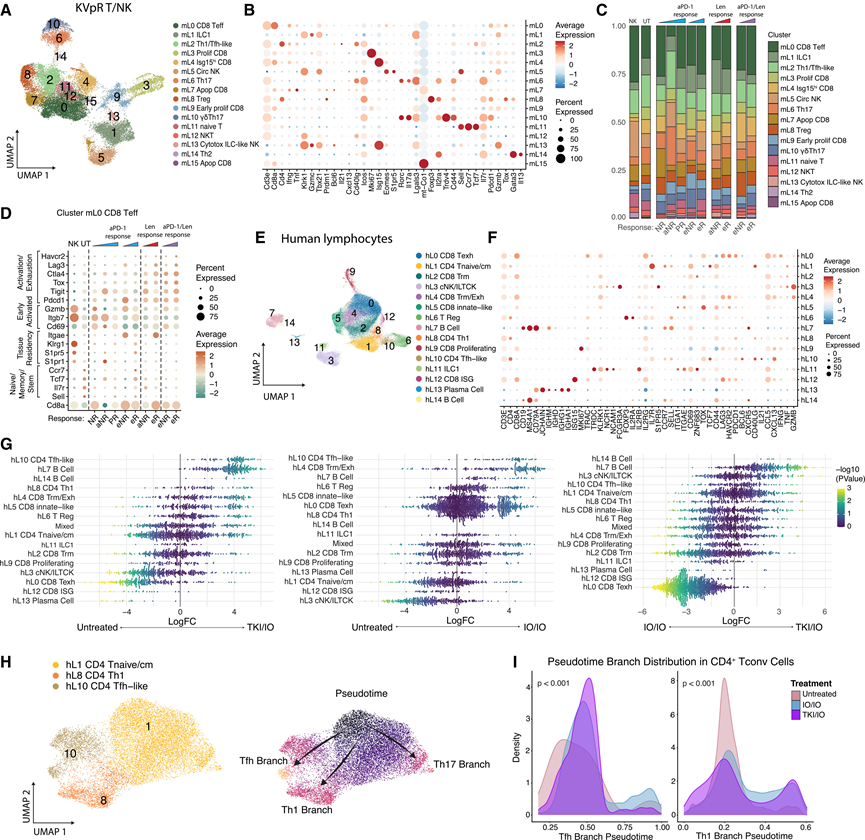
图5. TKI 和 IO 诱导不同的淋巴细胞反应。
(A-C) KVpR 肿瘤中 T 细胞和 NK 细胞的 ScRNA-seq UMAP、谱系定义基因的散点图和堆叠条形图。(D) mL0 CD8+ Teff聚类内功能基因的散点图。(E-F) 人类 scRNA-seq ccRCC 队列中所有淋巴细胞的谱系定义基因的 UMAP和散点图。(G) Milo 群图描绘了不同处理组之间差异细胞类型丰度变化。(H) CD4+ T 传统细胞类型的 UMAP (左),按沿三个 Palantir 伪时间分化轨迹的位置着色(右)。(I) Tfh 和Th1分支的核密度估计(KDE)按处理类型沿伪时间轴分布。
06
TKI/IO 治疗有效者的缺氧标志物存在空间相关性
他们使用接受治疗的人类ccRCC间质细胞 (IMC) 队列进行了正交验证(图 1A)。抗体组能够识别 14 种免疫细胞、基质细胞和肿瘤细胞群及其空间分布,其中混合谱系细胞归类为“其他免疫细胞”。与部分缓解 (PR) 患者相比,酪氨酸激酶抑制剂/免疫疗法 (TKI/IO) 完全缓解 (CR) 患者[包括接近完全缓解 (near-CR) 患者]的内皮细胞数量减少,但血管生成指数 (VN) 无差异;非缓解 (NR) 患者显示出类似的趋势,但样本量不足(图 6A-B)。相反,免疫疗法/免疫疗法 (IO/IO) 的缓解与内皮细胞丢失无关,虽然CR 患者的 VN 高于 PR 患者。
为了评估未接受匹莫尼达唑治疗的TME缺氧情况,他们检测了坏死和TME中HIF-1α的表达作为缺氧的替代指标。组织学坏死在接受TKI/IO治疗的患者中富集(10例注释病例中有8例),支持缺氧相关的机制,但由于组织核心数量有限,无法进行可靠的组间比较。低质量的坏死区域可能排除在TMA之外。有趣的是,中性粒细胞是HIF-1α的主要且最高表达细胞,其表达水平在图像缩放后超过了VHL缺陷型癌细胞。这使得他们能够选择性地评估TME缺氧,并将其与肿瘤假性缺氧区分开来,这与HIF-2α不同。HIF-1α的空间聚集(Moran's I)与内皮细胞比例呈负相关(图6 C),并且在存在坏死区域时与其共定位(图6 D),支持将其用作缺氧的替代指标。 GPNMB(一种 SPP1 TAM 的免疫细胞限制性标志物)的空间聚集(图 4 J)与 HIF-1α 聚集显著相关(图 6 E)。
接下来,他们对单核细胞、巨噬细胞和树突状细胞进行亚群分析,以鉴定GPNMB+和HLA-DR hi TAM聚类。他们证实了这些细胞群的预期功能二分性:与GPNMB+ TAM相互作用的CD8+ T细胞相比,与越来越多的HLA-DR hi TAM相邻的CD8+ T细胞表达更高的颗粒酶B和PD-1。与PR和NR患者相比,TKI/IO完全缓解(CR)患者中GPNMB+TAM的丰度呈现增加趋势(图6 F)。总而言之,TKI/IO治疗有效患者表现出协调的内皮细胞丢失、坏死以及空间聚集的HIF-1α+中性粒细胞和GPNMB+TAM(图6 G),这些特征在IO/IO治疗中并不存在,这支持了TKI在人ccRCC中以缺氧为中心的机制。
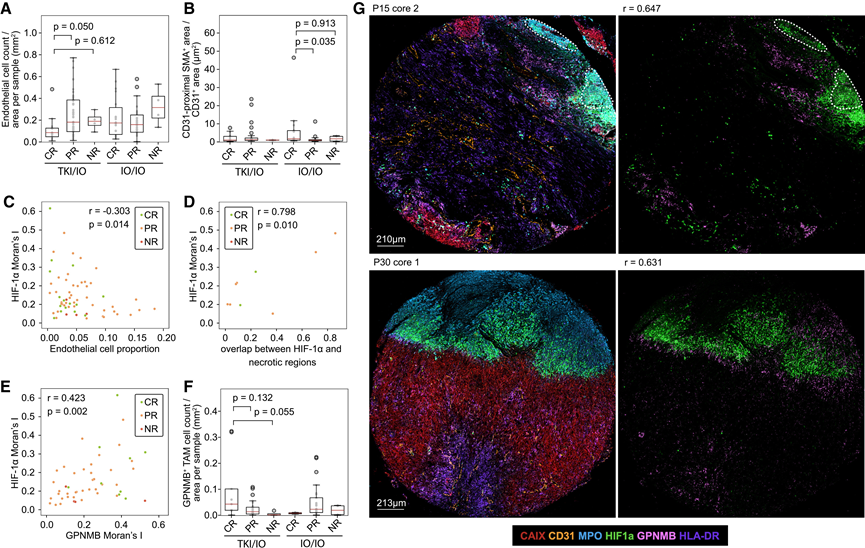
图6. TKI/IO 治疗有效者的缺氧标志物在空间上呈相关性。
(A-B) 人类 ccRCC 成像质谱流式细胞分析队列中血管生成和血管正常化的定量分析。(C-D) 仅在接受 TKI/IO 治疗的患者中,缺氧的公认特征与 HIF-1α Moran's I 之间的 Pearson 相关性:缺乏内皮细胞和与坏死重叠。(E) GPNMB Moran's I 与 HIF-1α Moran's I 在每个肿瘤核心中的关系。(F) 对不同治疗反应组中 GPNMB+ TAM 的丰度进行定量分析。(G) 具有高HIF-1α Moran's I 值和高GPNMB Moran's I 值的肿瘤核心代表性图像。
07
TKI/IO治疗可加剧小鼠的转移
鉴于缺氧与RCC和其他癌症转移之间的关联,他们探究了长期 TKI 治疗是否会促进转移扩散。由于 KVpR 模型的潜伏期较长,他们使用两种转移性同源ccRCC模型 LVRCC67和新近报道的 LVRCC88直接验证了这一点。这些模型均源自独立的肾脏肿瘤,通过 CRISPR 介导的Vhl、Trp53和Rb1基因缺失以及Myc 基因过表达而构建,并且与 KVpR 和人类基质/增殖性 ccRCC 高度相似。在皮下 (SC) LVRCC88 模型中,肿瘤最初对Len敏感,但很快产生耐药性(图7A -B)。值得注意的是,与对照组相比,Len 还增加了肺和肝转移,而添加抗 PD-1 抗体进一步加剧了该模型中的转移负荷(图 7 C)。免疫荧光染色证实了Len诱导的缺氧和血管丢失,但由于坏死区域与组织切片的粘附性较差,缺氧程度可能低估(图7D-E)。SC LVRCC67肿瘤对Len也表现出类似的短暂性、非治愈性反应(图7F)。Len同样增加了该模型中的转移负荷,而抗PD-1抗体并未进一步增强转移,且与生存偏倚无关(图7G-H)。与缺氧增加和血管修剪一致,流式细胞分析显示Len治疗后TAM浸润增加和SPP1表达升高(图7I)。观察与mM0抗原呈递TAM相关的标志物,他们发现虽然MHC II类分子水平没有差异,但CD206在Len治疗后也升高(图7I),提示同时存在缺氧和炎症性重塑。此外,Len 还诱导了 cDC1 的显著扩增,这与 KVpR 的研究结果一致,而 B 细胞、中性粒细胞和 T 细胞的预期变化较弱或不一致,这可能反映了组织处理方式的差异。在原位 LVRCC88-M1 肿瘤中,Len 的反应是短暂的(图 7 J-K),但尽管原发肿瘤耐药,转移负荷仍然升高(图 7 L)。这些肿瘤表现出缺氧增加(图 7 M-N),但不如 SC 肿瘤明显,且没有明显的血管消融(图 7 N),这与血管异常而非丢失相符。相比之下,肝转移灶对 Len 诱导的缺氧和坏死仍然敏感。静脉注射转移性 LVRCC67 衍生物优先播散至肝脏,并有 Len 增强转移负荷的趋势,这共同表明 TKI 诱导的缺氧应激可能选择来自原发部位和转移部位的转移克隆。
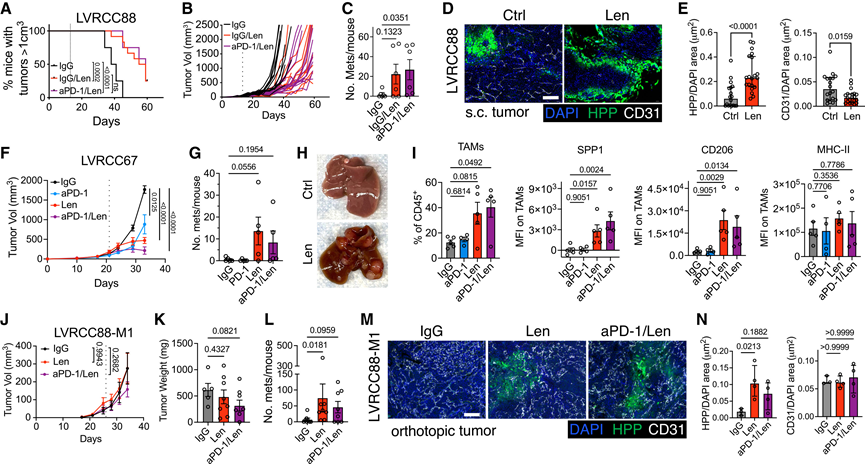
图7. TKI 诱导的缺氧促进同源 ccRCC 模型中的转移。
(A) 原发肿瘤体积阈值的Kaplan-Meier曲线。(B) 单个肿瘤体积和(C)转移灶计数。(D)代表性肿瘤免疫荧光染色(E)定量分析。(F) 肿瘤体积。(G)转移计数。(H)转移结节的代表性图像以及(I)通过流式细胞技术进行的 TAM 分析。(J)平均肿瘤生长量,(K)终点时的肿瘤重量。(L)转移负荷。(M)代表性原发肿瘤的免疫荧光(IF)。(N)定量分析。
08
缺氧和免疫激活可预测对TKI/IO的反应
系统性免疫和代谢特征与RCC中酪氨酸激酶抑制剂/免疫疗法(TKI/IO)的疗效相关,可能反映了肿瘤缺氧和TAM驱动的免疫抑制。因此,他们评估了来自VEGFR-TKI/IO(JAVELIN Renal和MSKCC RWD)和抗VEGF抗体/免疫疗法(IMmotion151/150)试验(统称为VEGF/IO)的基线肿瘤组织中scRNA-seq衍生的髓系特征,作为潜在的生物标志物。为了捕捉缺氧和免疫激活之间的平衡,他们生成了一个综合的mM1_mM0_Diff评分,即从缺氧的mM1 TAM特征中减去免疫激活的mM0(评分越高反映缺氧程度越严重),并将其与已建立的预测因子进行比较,包括血管生成、JAVELIN_26(细胞毒性淋巴细胞特征)和MSK-I(腺苷和髓系特征的综合指标)。
在 JAVELIN Renal 101、IMmotion151 和 IMmotion150 试验中,mM0 特征在任何治疗方案中均不具有预测价值(图 8A-C),这可能反映了抗原呈递 TAM 的双重作用。mM1 特征与 VEGF/IO 治疗的不良预后趋势相关,但未达到统计学意义。相反,mM1_mM0_Diff 评分显著预测了 JAVELIN Renal 101 和 IMmotion150 试验中 VEGF/IO 治疗的较差反应,表明基线缺氧程度高且促炎信号传导低的肿瘤对联合治疗的反应较差。相反,mM14 cDC1 特征在所有队列中均与 VEGF/IO 治疗反应的改善相关,其表现与 JAVELIN_26 相当,并支持了预先存在的抗肿瘤免疫状态的重要性。在 IMmotion150 的 IO 单药治疗组中,较高的 mM1 和 mM1_mM0_Diff 评分也与阿特珠单抗疗效不佳相关(图 8C)。在舒尼替尼(第一代 VEGFR-TKI)单药治疗组中,血管生成如预期般预测了疗效,而缺氧相关特征(mM1、mM1_mM0_Diff)则预测了不良预后。
接下来,他们在一个富含当代TKI/IO方案(特别是乐伐替尼/帕博利珠单抗)的真实世界ccRCC队列中评估了这些特征(MSKCC RWD)。较高的基线mM1_mM0_Diff表达与较差的总生存期(OS)相关(图8D),证实了缺氧、免疫抑制肿瘤对TKI/IO的耐药性。与试验队列不同,较高的mM0_SelenopTAM和JAVELIN_Angio表达预示着OS的改善,这可能反映了基于乐伐替尼联合疗法的不同机制,但仍需进一步验证。近期来自Len/帕博利珠单抗3期试验的生物标志物分析并未发现缺氧或糖酵解特征具有预测价值;然而,这些分析与SPP1+ TAM特征重叠甚少,且未报告OS。
这些数据共同表明,虽然治疗引起的缺氧伴随着免疫激活和有利的VEGF/IO反应,但基线缺氧伴低免疫浸润预示着单药治疗和联合治疗方案的疗效均较差(图8E)。这种看似矛盾的现象可能反映了不同的生物学背景:基线缺氧与不受控制的增殖和免疫通路不良相关,而急性治疗引起的缺氧则反映了血管塌陷和肿瘤死亡。
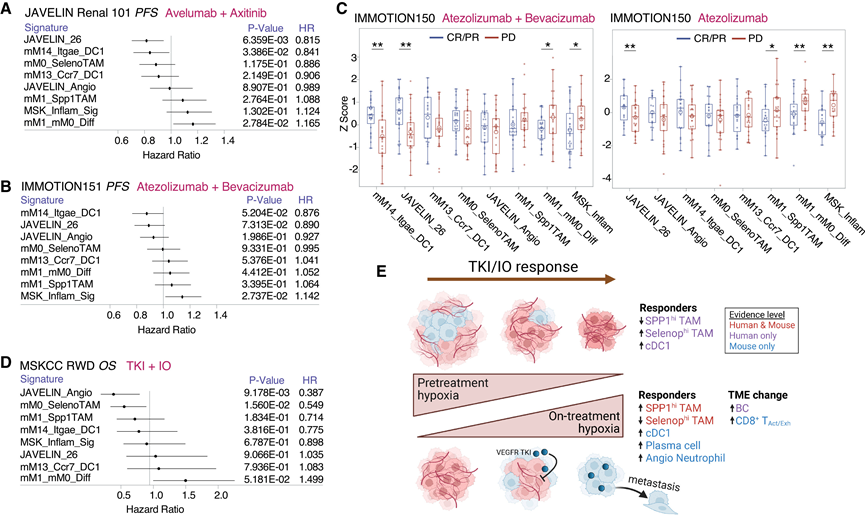
图8. 缺氧和炎症预测 TKI/IO 反应。
(A-B) JAVELIN Renal 101 和 IMmotion151 试验中 VEGF/IO 组的基因特征森林图。(C) 箱线图显示了IMmotion150队列中VEGF/IO组或仅IO组按放射学反应分组的基因特征表达情况。(D) MSKCC RWD VEGFR-TKI/IO队列的森林图。(E) 缺氧悖论模型的概要示意图。
+ + + + + + + + + + +
结 论
本研究利用转基因ccRCC小鼠模型和多组学技术,研究了VEGFR-TKI、aPD-1以及VEGFR-TKI/aPD-1联合治疗后肿瘤微环境的演变。在基线假性低氧肿瘤中,存在少量对低氧敏感的SPP1+ TAM。这种真实缺氧的替代指标与小鼠和人类接受 VEGFR-TKI/aPD-1 治疗后成功反应相关,反映了治疗诱导的缺氧性坏死。然而,矛盾的是,在 VEGFR-TKI/aPD-1 临床试验和真实世界队列中,治疗前缺氧预示着更差的预后,而延长暴露于诱导缺氧的 VEGFR-TKI 则会加剧小鼠的转移,这凸显了缺氧在 ccRCC 疾病进程中的双重影响。
+ + + + +



