

 English
English文献解读 |Sci Adv ( 12.5 ):粪便代谢物分析可鉴定 30 天死亡率较高的危重患者
✦ +
+
论文ID
原名:Fecal metabolite profiling identifies critically ill patients with increased 30-day mortality
译名:粪便代谢物分析可鉴定30天死亡率较高的危重患者
期刊:Science Advances
影响因子:12.5
发表时间:2025.06.06
DOI号:10.1126/sciadv.adt1466
背 景
危重症患者常诊断患有急性呼吸窘迫综合征 (ARDS) 或脓毒症等综合征。这些综合征的发病机制存在异质性。这种生物学异质性导致临床试验中对治疗的反应各不相同。精准医疗的干预目标是针对生物学状态而非综合征,并有可能改善治疗反应。精准医疗的目标是促进对可治疗性状的因果干预。多项研究将可治疗性状定义为一种特定的生理紊乱,其特征是生物标志物,这些生物标志物预示着对特定疗法的可预测反应。入住重症监护病房 (MICU) 的危重症患者肠道菌群多样性降低,且菌群相关代谢物浓度发生改变。肠道菌群产生的代谢物与接受复杂治疗的患者的生存期相关,因此可能是一种可治疗的特性,可改善临床预后。
实验设计
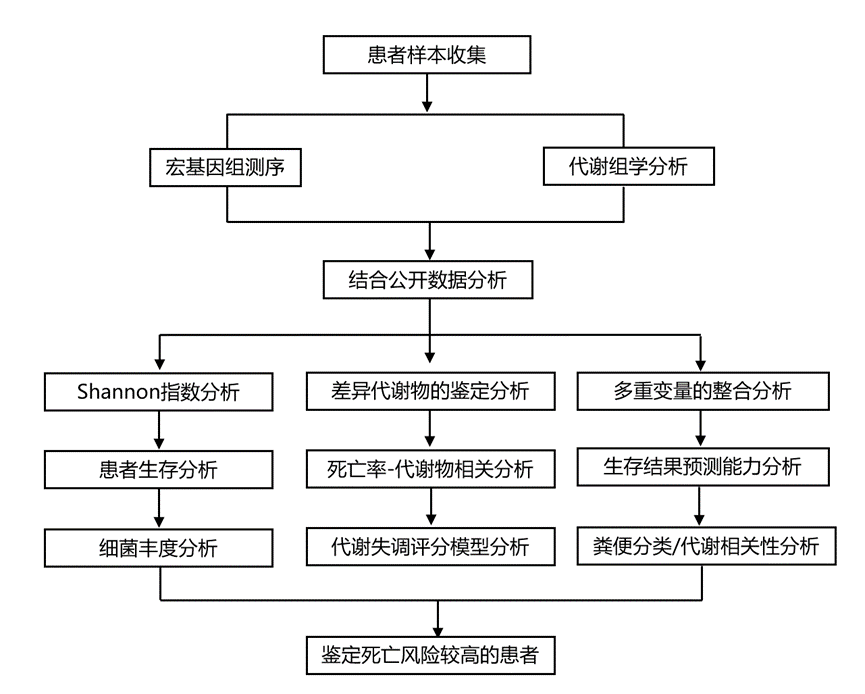
结 果
01

患者样本的多组学分析
他们从治疗前肿瘤活检样本中收集了多组学数据(图 1B)。具体而言,对 104 对肿瘤和血液样本进行了全外显子组测序 (WES),以鉴定癌症基因组中的体细胞单核苷酸变异 (SNV)、小片段插入/缺失 (INDEL) 和体细胞拷贝数变异 (CNV)。此外,还分别对 145 份、140 份和 137 份肿瘤样本进行了全基因组亚硫酸氢盐测序 (WGBS)、转录组分析(RNA-seq) 和液相色谱-质谱分析,以分析甲基化组、转录组和蛋白质组/磷酸化组数据。在所有入组患者中,89 例拥有上述所有组学平台的数据。
使用GATK Mutect2对所有样本进行体细胞SNV和小INDEL检出,编码区非同义突变负荷中位数为74个(范围从3到1078个)。这些突变包括先前报道的乳腺癌驱动基因,例如TP53、PIK3CA、KMT2D、KMT2A和KMT2C、GATA3、MED13和CSMD1(图2A)。这些驱动基因的中位变异等位基因频率为 0.166。突变特征分析表明,年龄相关特征(SBS1 和 SBS5)和 APOBEC(载脂蛋白 B mRNA 编辑酶,催化多肽样)特征(SBS3 和 SBS13)占主导地位,这表明这些过程驱动了大部分观察到的诱变(图 2B)。正如预期的那样,SBS1 和 SBS5 与患者诊断时的年龄呈正相关。值得注意的是,SBS3 特征在 ER− HER2−中富集,但在 SBS13 中没有富集。随后,应用癌症重要靶点基因组识别2 (GISTIC2) 在该队列中发现了复发性局灶性和臂水平拷贝数变异。与先前结果一致, ERBB2、MYC、AIM1和CCND1的扩增在乳腺癌队列中大量存在(图2B)。
为了探究体细胞突变是否有助于治疗反应,他们比较了 pCR 组和 RD 组中非静默体细胞突变的频率。在校正错误发现率 (FDR) 后,体细胞基因 SNV/INDEL 与 pCR 无显著相关性。对于 CNV,通过比较各亚型中 pCR 组和 RD 组的局部 CNV 分布,他们发现ER− HER2 −患者群体中 RD 组的6q27拷贝数显著较低(图 2C-E)。进一步纳入更多样本或许可以验证这一观察结果,并能够识别更多可预测治疗结果的基因变异。
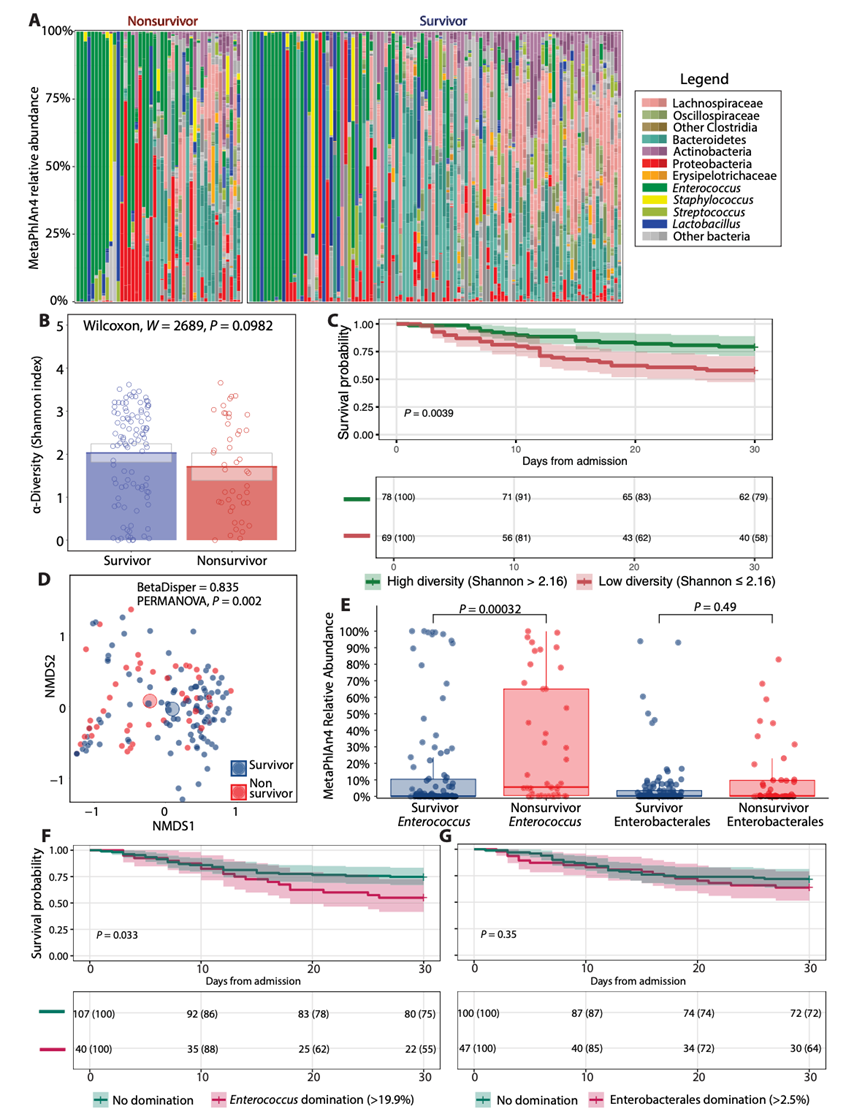
图1.鸟枪法宏基因组数据衍生的多样性指标和与生存结果相关的分类单元。
(A) 每位患者的相对丰度,其中分类单元按不同的、生物相关的分类学水平分组和着色。(B) alpha 多样性条形图。(C) 入住 ICU 后 30 天生存概率的 Kaplan-Meier 曲线。(D) 由非度量多维尺度 (NMDS) 确定的 beta 多样性。(E) 致病分类单元相对丰度的箱线图。(F) 入住 ICU 后 30 天生存概率的 Kaplan-Meier 曲线,根据肠球菌相对丰度状态(属级)对患者进行分层。(G) 入住 ICU 后 30 天生存概率,根据肠杆菌目相对丰度状态(目级)对患者进行分层。
02
单一代谢物与 30 天死亡率之间无关联
微生物群会产生大量代谢物,这些代谢物会影响人类健康。因此,他们研究了幸存者和死亡者之间粪便代谢物浓度的差异。他们对 83 种粪便代谢物进行了相对定量,包括 SCFA、初级和次级胆汁酸以及色氨酸代谢物(包括吲哚)(图 2A)。幸存者和死亡者的粪便浓度没有显著差异。然而,与死亡者相比,幸存者中的 13 种代谢物(全部是胆汁酸)具有显著变化。与死亡者相比,幸存者中的色氨酸、牛磺石胆酸和熊去氧胆酸具有显著差异。
他们还对30种代谢物进行了绝对定量分析,包括短链脂肪酸(SCFA)、初级胆汁酸和次级胆汁酸、氨基酸以及色氨酸代谢物(吲哚、血清素及其衍生物、犬尿氨酸及其衍生物)。单变量分析显示,幸存者和死亡者粪便中这些代谢物的浓度无显著差异(图2B)。综上所述,这些结果表明幸存者和死亡者之间单一代谢物浓度没有显著差异。然而,幸存者粪便中次级胆汁酸浓度呈升高趋势。
他们推断,整合粪便代谢物浓度的综合模型可能与 30 天死亡率相关。粪便代谢失调评分 (MDS)由三种次级胆汁酸(脱氧胆酸、石胆酸和异脱氧胆酸)和脱氨酪氨酸组成,可预测 COVID-19 危重患者的死亡率并与其独立相关。在非 COVID-19 MICU 患者队列中,与低分(<2 分)患者相比,高分(≥2 分)患者死亡率更高,如 Kaplan-Meier 生存曲线所示。尽管 COVID-19 MDS 对正确分类当前队列中的非幸存者具有很高的特异性,但准确度和灵敏度较低。包含COVID-19 MDS及其混杂因素(CCI和SOFA)的Cox比例风险模型符合比例风险假设,但与30天死亡风险增加无关。这表明COVID-19 MDS可能不适用于非COVID-19 MICU患者。
他们使用绝对粪便代谢物浓度建立了逻辑回归、极端梯度提升和随机森林模型来预测死亡率。此外,还建立了一个基于粪便代谢物浓度最佳阈值(以毫摩尔为单位)的 MDS 模型,该阈值是通过优化每种代谢物的接收者操作特性 (ROC) 分析中的约登指数确定的。方向是相对于非存活组确定的。为了评估每种代谢物的变量重要性,进行了岭回归以预测生存结果。然后,根据代谢物的变量重要性,使用岭回归确定的 β 系数,按顺序将其纳入 ROC 分析,直到使用完所有代谢物(图 3)。吲哚-3-乙酰胺未纳入模型,因为只有四个值高于检测限(图 2B)。与 COVID-19 MDS 类似,对于每位患者,得分是通过为与死亡幸存者相关的浓度范围内的每种代谢物打分来确定的。所有分数的总和就是 MDS。再次使用约登指数计算 MDS 的阈值。对于每种代谢物组合,评估模型性能指标——准确度、特异性、敏感性和限制平均生存时间 (RMST) 差异。选择了最简单但性能最佳的模型,该模型由 13 种代谢物组成(图 3)。阈值为 7.5,这意味着得分 > 7.5 代表严重的代谢失调。
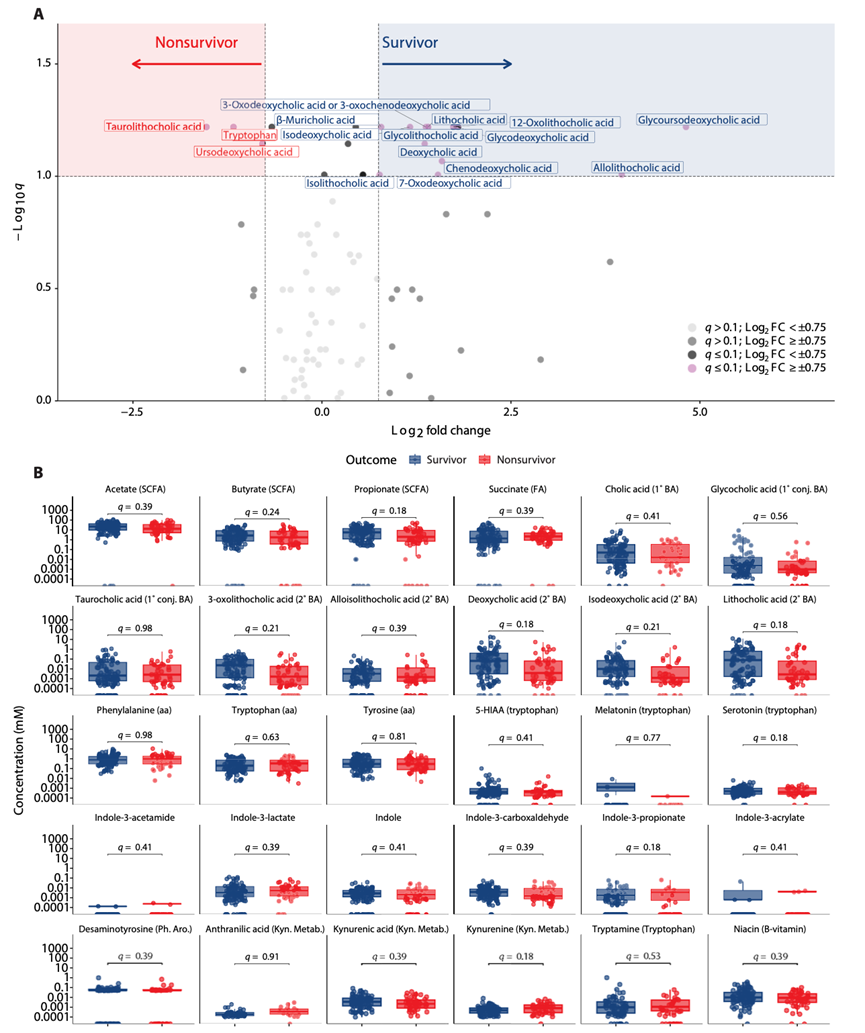
图2. 幸存者和非幸存者的定性和定量代谢组学。
(A) 对定性估计代谢物的火山分析。(B) 对存活组的代谢物进行定量计算。
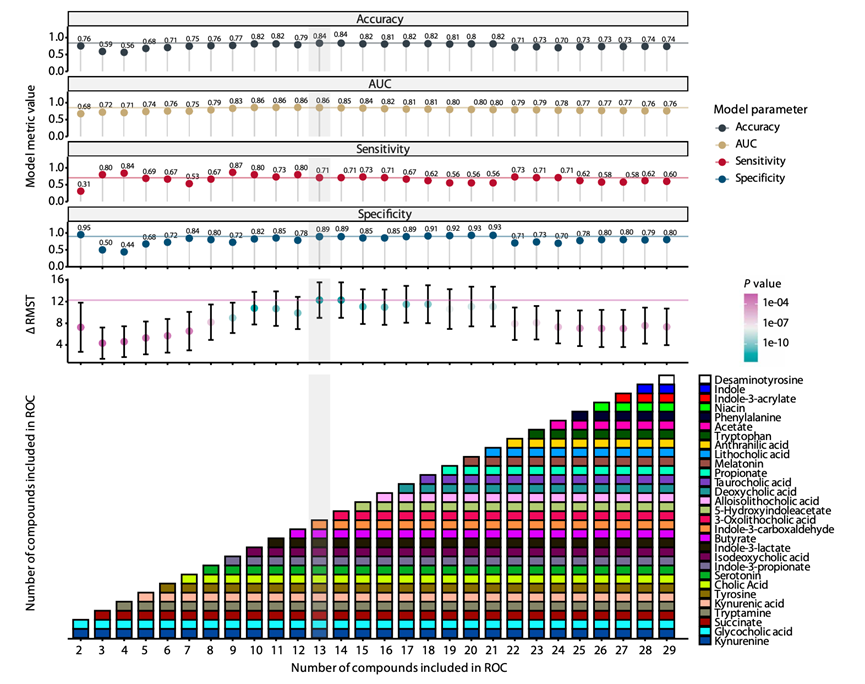
图3. MDS 模型和生存指标。
03
MDS 可预测 30 天死亡率,且与 30 天死亡率独立相关
在验证队列中,随机森林和 MDS 模型具有相当的准确度、灵敏度和特异性,并且优于 COVID-19 MDS。与随机森林和 MDS 模型相比,逻辑回归和极端梯度增强模型缺乏灵敏度。MDS 模型中包含的 13 种代谢物与随机森林模型最重要的代谢物大量重叠,这由它们的基尼重要性决定。他们选择继续使用 MDS,因为它是性能最佳的模型之一,具有更高的可解释性,可以进行组间比较。此外,MDS 还能阐明与 30 天死亡率可能相关的代谢物浓度。在训练队列中,非幸存者的 MDS 显著高于幸存者(图 4A)。Kaplan-Meier 生存曲线显示,低 MDS(评分 < 7.5)患者与高 MDS(评分≥7.5)(图 4B)。在验证了比例风险假设后, MDS 每增加一个单位,在控制混杂因素后,30 天死亡风险就会增加 1.73 倍。当将 MDS 作为二分变量纳入 Cox 比例风险模型时,他们发现基于 MDS 的代谢失调≥7.5 的数据显示30 天死亡风险增加 8.66 倍。在将分析限制在 ICU 入院后 3 天内收集的粪便样本(占总患者的 65%)后,训练队列中的生存分析和 Cox 比例风险模型显示出相似的结果。在这些患者中,MDS 模型的准确度为 0.84,特异性为 0.75。
在验证队列中,幸存者和死亡患者的 MDS 没有显著差异(图 4C),尽管死亡患者的平均值较高。Kaplan-Meier 生存曲线显示,低 MDS(评分 < 7.5)患者与高 MDS 患者的生存概率存在显著差异(图 4D)。在 Cox 比例风险模型中,他们发现 MDS 每增加一个单位,30 天死亡风险就会增加 1.21 倍,但是这并不显著(P = 0.289)。当将MDS纳入二分变量时,代谢失调会使30天死亡风险增加1.88倍,差异不显著(P =0.291)。验证队列的结果显示出与训练队列相同的趋势,但由于样本量较小,无法证明显著性,因此需要在更大的独立队列中验证MDS。
综上所述,MDS是一个由13种粪便代谢物组成的评分系统,可以准确预测异质性危重症患者队列的30天死亡率。与30天死亡率的独立关联表明,MDS中的代谢物是潜在的候选干预因素(即可治疗的特征),可以改善危重症患者的预后,但尚需进一步验证。
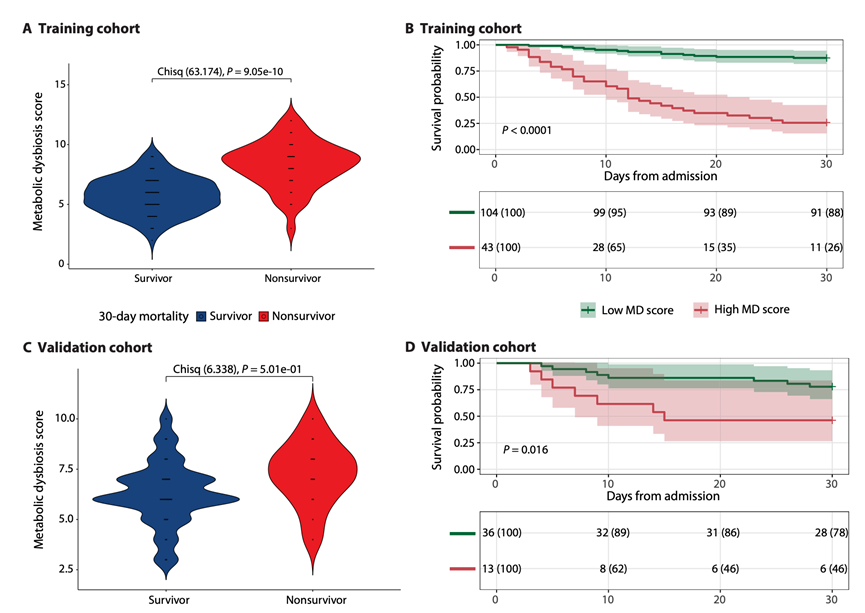
图4. 评估 MDS 对训练和验证队列的生存结果预测能力。
(A,C) 训练队列和验证队列中幸存者和死亡患者的 MDS 小提琴图。(B, D) 根据训练队列和验证队列的 MDS(由最佳阈值分析确定)对患者进行分层。
04
分类参数不能准确反映粪便代谢失调
在训练队列中,α多样性与 MICU 入院后 30 天的死亡率无关,而代谢失调(定义为 MDS > 7.5)则与此相关。虽然低和高 MDS 的患者在分类多维尺度图上聚集在一起(图 5A),但 24.5% 的患者具有低 Shannon 多样性指数但未处于代谢失调状态,6.8% 的患者具有高 Shannon 多样性指数但处于代谢失调状态(图 5B)。此外,虽然 Shannon 指数和 MDS 之间存在显著相关性,但这种相关性是中等(图 5C)。使用MaAsLin2进行差异丰度分析(未校正协变量)表明,Evtepia、Lachnospiraceae和 Oscillospiraceae的成员与低MDS显著相关(图5D)。然而,当校正协变量后,这些关联不再显著。先前已证实粪便代谢物浓度与微生物组代谢基因丰度之间缺乏相关性,这表明营养物质的有效性等外源因素会影响MDS(21)。总之,分类学参数与粪便代谢失调的程度无关。
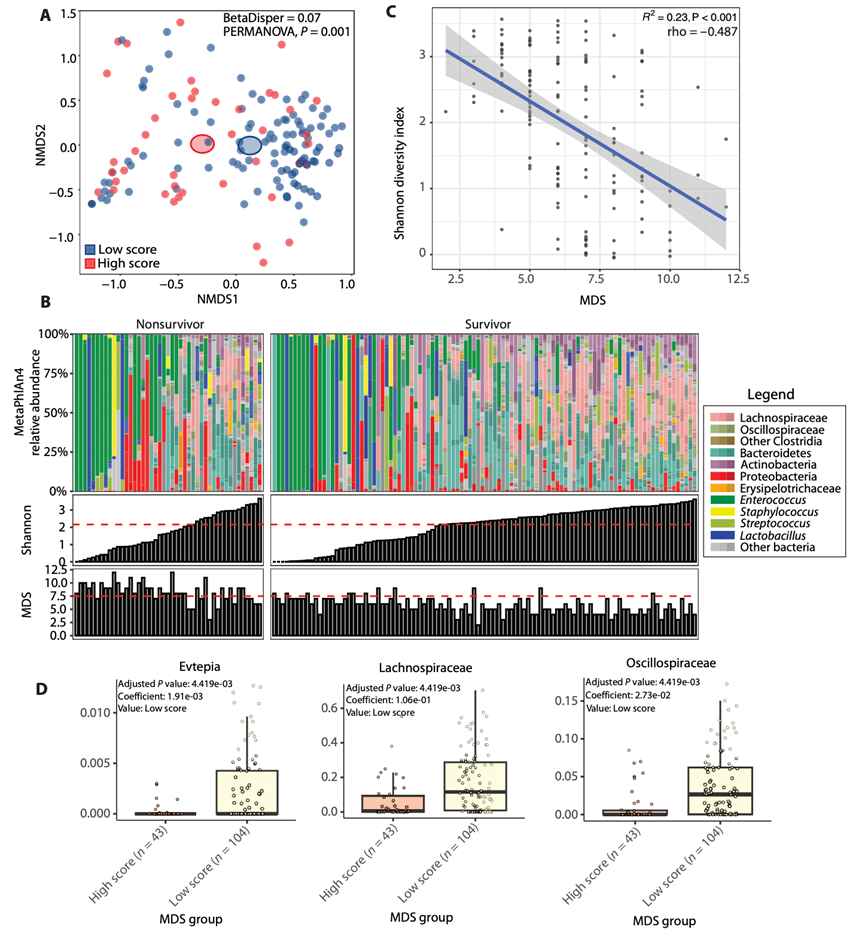
图5. 粪便分类和代谢相关性。
(A) 基于 Bray-Curtis 距离度量的非度量多维尺度图。(B) 上图显示了每位患者的相对丰度,其中分类单元按不同的生物学相关分类学水平分组和着色。下图分别显示了每位患者的香农多样性指数和 MDS。(C) 该图显示了y轴上的香农多样性指数与x轴上的 MDS之间的相关性。(D) 箱线图显示了未校正混杂因素的科级 MaAsLin2 差异丰度分析结果。
+ + + + + + + + + + +
结 论
本研究前瞻性地收集了196例因非COVID-19呼吸衰竭或休克入住MICU的危重症患者的粪便样本,通过宏基因组测序确定了微生物组组成,并通过质谱法定量了微生物组来源的粪便代谢物,以将微生物组特征和代谢物与30天死亡率关联起来。入住MICU后第一份粪便样本的微生物组组成与30天死亡率无独立相关性。本研究开发了一个代谢失调评分 (MDS),该评分使用13种微生物组来源代谢物的粪便浓度,可预测30天死亡率,且不受已知混杂因素的影响。 MDS 补充了现有工具,通过结合潜在可修改的、与微生物组相关的、独立的宿主恢复力贡献者来筛选具有高死亡风险的患者。
+ + + + +



