

 English
English文献解读|Nat Commun(15.7):肾母细胞瘤的综合蛋白质组学表征
✦ +
+
论文ID
原名:Integrative proteogenomic characterization of Wilms tumor
译名:肾母细胞瘤的综合蛋白质组学表征
期刊:Nature Communications
影响因子:15.7
发表时间:2025.08.19
DOI号:10.1038/s41467-025-62234-7
背 景
肾母细胞瘤(Wilmstumor,WT)是儿童中最常见的恶性肾脏肿瘤类型,约占所有肾脏肿瘤的 90% 和所有儿童恶性肿瘤的 7% 。东亚地区 WT 的年发病率为每百万 4.3 例,低于北美或欧洲(每百万 8-9 例)。在中国 0-4 岁儿童中,该恶性肿瘤的发病率高于每百万 7 例。随着规范化治疗方案的实施,WT 的总体生存率已得到显著提高,但对于组织学类型不佳的患儿,4 年生存率为 30%-85%,具体取决于肿瘤分期。目前,约有40个体细胞突变或拷贝数(CN)变异是WT肿瘤发生和发展中的关键癌症驱动候选基因,包括WT1、CTNNB1、AMER1、IGF2、TP53和MYCN。WT 中最突出的三个遗传特征和表观遗传改变是WT1的功能丧失、Wnt信号通路的激活和IGF2的过表达。然而,WT中基因突变和CN变异的频率远低于成人肿瘤,这在一定程度上阻碍了针对WT儿童的靶向治疗的开发。
实验设计
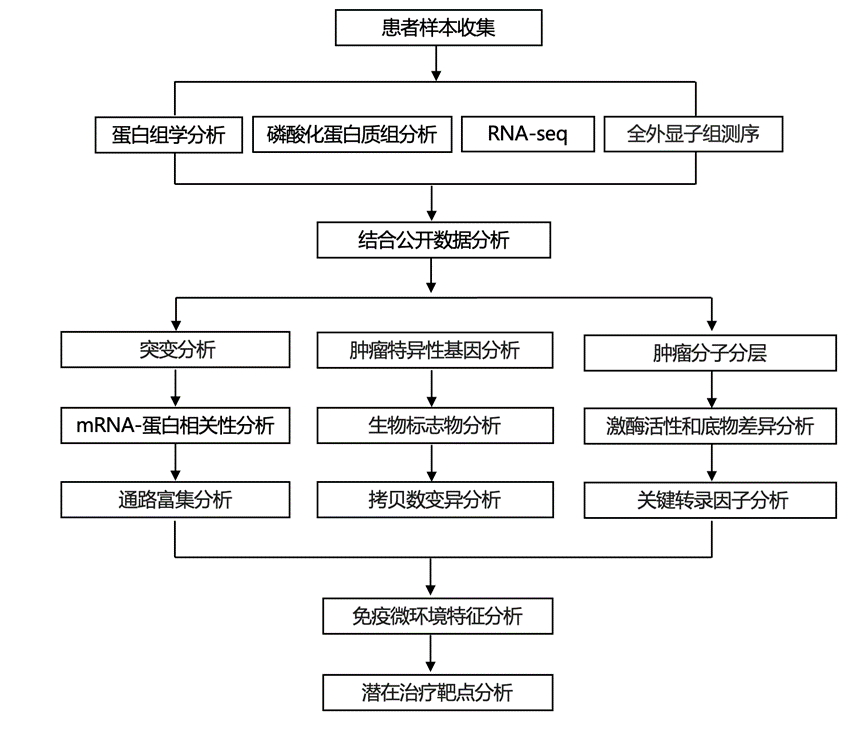
结 果
01

WT肿瘤队列的研究设计和多组学结果
本研究招募了96例18岁前确诊为WT的患者。该队列包含91例WT样本和74例NAT样本。为了全面分析WT的分子特征,研究团队对这些样本进行了全外显子组测序(WES)、RNA测序(RNA-seq)、定量蛋白质组学和磷酸化蛋白质组学分析(图 1a-c)。
在 36 个 WT 样本中,他们总共鉴定出 369 个非沉默突变,包括 317 个替换(291 个错义突变、18 个无义突变、7 个剪接突变和 1 个翻译起始位点突变)和 52 个插入/缺失(21 个移码突变、29 个移码内突变、1 个剪接突变和 1 个无义突变),导致肿瘤突变负担 (TMB) 中等为每百万碱基 0.15 个。他们还从62个WT和37个正常癌旁组织(NAT)样本中鉴定出总共16835个编码蛋白质的RNA,从88个WT和71个NAT样本中鉴定出9956个蛋白质,从23对WT和NAT样本中鉴定出9343个磷酸化位点,这些位点位于2918个蛋白质中。在磷酸化位点中,8703个(93.15%)是从Signor或PhosphoSite数据库中整理出来的,这表明本研究的磷酸化蛋白质组数据具有很高的可靠性。值得注意的是,与NAT相比,WT中鉴定的基因,磷酸化位点和磷酸化蛋白质的数量显著更高,这表明肿瘤细胞经历了基因表达和蛋白质活性的复杂改变(图 1d)。
此外,他们对转录组学和蛋白质组学数据鉴定的mRNA-蛋白质对进行了相关性分析。具体而言,他们发现WT样本的Spearman相关性高于NAT样本(图 1e)。转录组和蛋白质组之间的不一致性也暗示了转录组无法观察到的额外信息。在WT样本中,参与CDK调控DNA复制的mRNA /蛋白质和控制肾脏发生的基因呈正相关(图 1e)。呼吸电子传递,IL-2信号通路和脂肪酸代谢等途径的mRNA /蛋白质水平在NAT样本中呈正相关(图 1e)。这些结果表明,WT 样本中的细胞增殖、DNA 复制和肾脏发育比 NAT 样本表现出更高的活性。
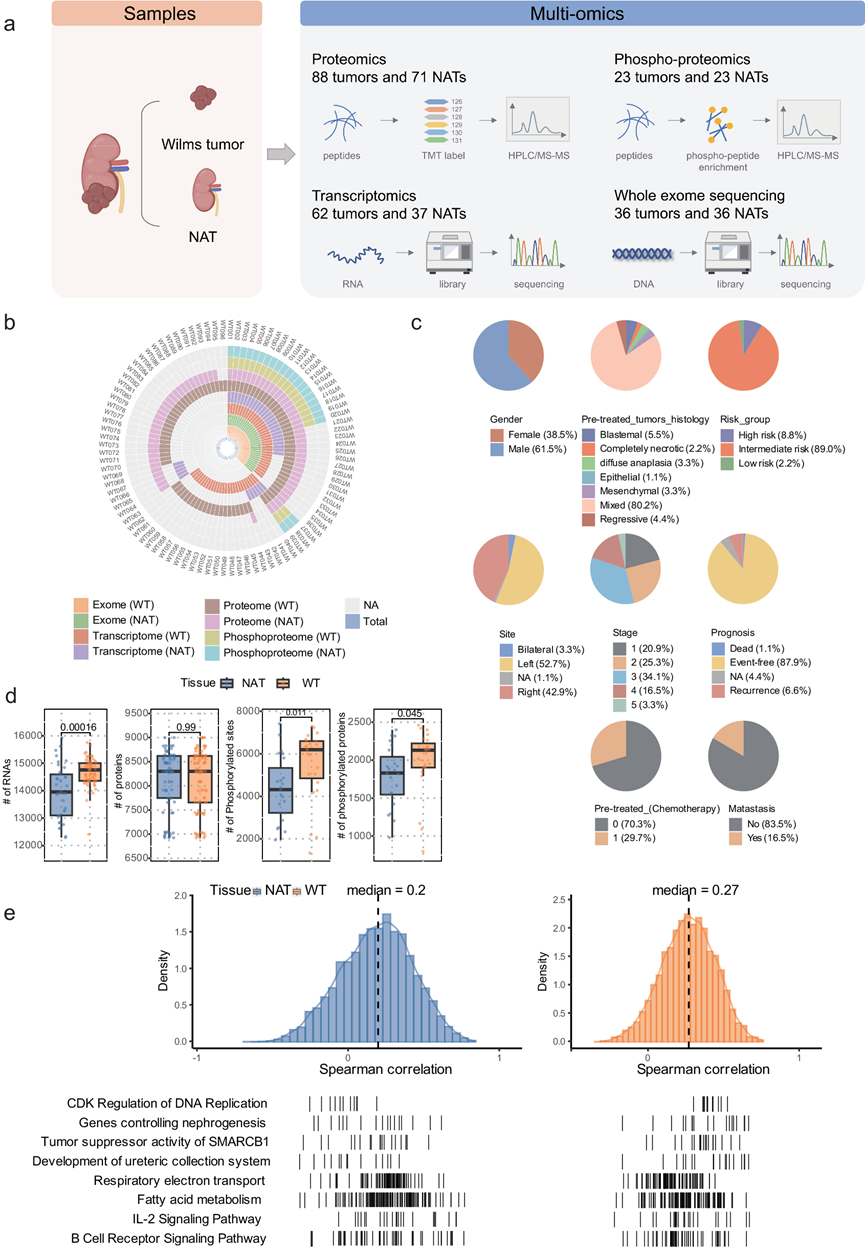
图1. WT 肿瘤队列的研究设计和蛋白质组学概况。
(a) 肿瘤多组学分析的实验设计和概述。(b) 以患者为中心的 Circos 图。(c) 饼图显示了本研究中基于关键临床和预后变量的 WT 队列内患者分布。(d) 箱线图显示了通过多组学分析鉴定的 WT 和 NAT 中 RNA、蛋白质、磷酸化位点和磷酸化蛋白质的数量分布。(e) NAT(左)和 WT(右)样本中基因级 mRNA-蛋白质相关性。
02
肿瘤-NAT比较揭示了致瘤基因和潜在的生物标志物
他们在mRNA水平上鉴定了WT组织中6174个上调基因和3714个下调基因。此外,在WT组织中发现了1827个上调和2240个下调的蛋白质。对于磷酸化蛋白质组数据,他们在质量控制后保留了9343个磷酸化位点以供进一步分析,从而鉴定了WT组织中1056个上调和530个下调的磷酸化蛋白质。此外,通过转录组、蛋白质组和磷酸化蛋白质组数据,在三个表达水平上进行差异表达分析,共鉴定出351个上调的基因产物(图 2a-b)。每个组学数据集都揭示了一组独特的差异表达基因产物,凸显了多组学整合分析在癌症研究中的重要性。
差异表达分析还发现,一些WT的诊断生物标志物在WT样本中也在mRNA和蛋白质水平上过表达(图 2c)。接受(化疗样本)或未接受(治疗初治样本)化疗的WT样本均纳入WT与NAT的差异分析中,相关性分析显示,接受或未接受化疗的WT样本与NAT相比,差异表达基因的一致性较高。就差异基因表达的程度而言,治疗初治组中上调的蛋白质在化疗组中减少,但相对于NAT仍然过表达。
差异表达的mRNA、蛋白质和磷蛋白的功能富集分析表明,与细胞增殖和细胞周期(细胞周期、有丝分裂中期和后期以及癌症中的视网膜母细胞瘤基因)、RNA加工(mRNA剪接和RNA聚合酶II转录终止)、表观遗传调控(ERCC6(CSB)和EHMT2(G9a)正向调节rRNA表达、基因表达的表观遗传调控)、肾脏发育(输尿管集合系统的发育)、NOTCH和Wnt信号传导相关的通路在WT中上调的mRNA、蛋白质和磷蛋白高度富集,表明它们在WT中具有潜在的高活性并且与WT的肿瘤发生密切相关(图 2d)。相反,正常代谢(氨基酸代谢、糖酵解/糖异生和脂肪酸代谢)、肾功能(近端小管转运)和基质(细胞-细胞通讯、白细胞跨内皮迁移、粘着斑)因下调的mRNA、蛋白质和磷蛋白而富集,表明WT的正常代谢能力和肾功能受损(图 2d)。此外,他们还发现与免疫反应相关的途径(补体级联、FCGR激活、CD22介导的BCR调节以及抗原加工和呈递)因下调的mRNA或蛋白质而特异性富集(图 2d),表明WT中免疫相关过程可能受到抑制或失调。具体而言,这些途径中涉及的关键基因在WT和NAT样本之间的RNA和蛋白质水平上均存在差异表达(图 2e)。
此外,对差异磷酸化蛋白和磷酸化位点的分析揭示了几个导致这些磷酸化变化的关键激酶。值得注意的是,他们观察到WT中ATM、CDK1和CDK2等激酶活性增加,这些激酶可能是WT中潜在的治疗靶点(图 2f)。
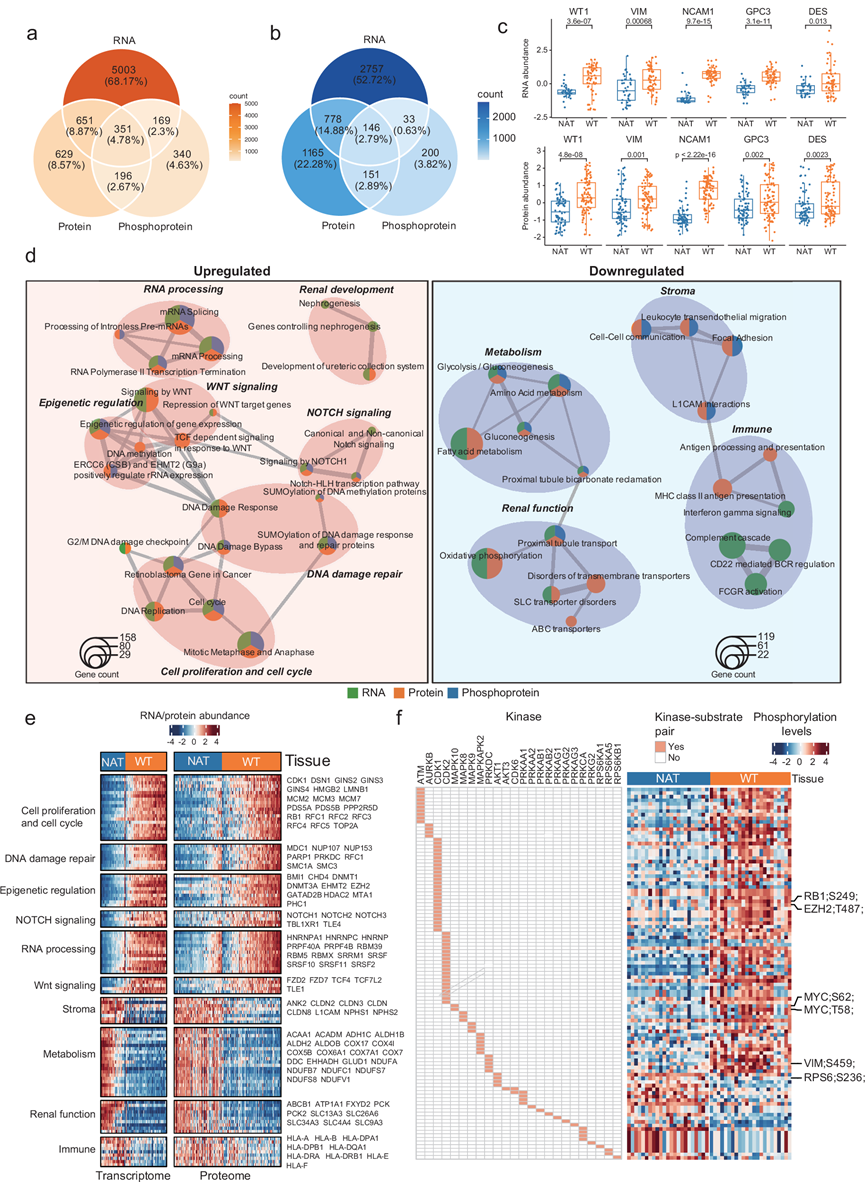
图2. WT 中肿瘤特异性基因和通路的识别。
(a) 维恩图显示了 WT(Wilms 肿瘤)中 RNA、蛋白质和磷酸化蛋白的基因上调重叠情况。(b) 维恩图显示了 WT(Wilms 肿瘤)中 RNA、蛋白质和磷酸化蛋白的基因下调重叠情况。(c) 箱线图显示了 WT和 NAT中所示基因的 Z 分数标准化 RNA(上图)和 蛋白质(下图)丰度 。(d) 与NAT 相比,WT 中上调(左)和下调(右)的 RNA/蛋白质/磷酸化蛋白质富集的通路。(e) WT 和NAT之间差异表达RNA/蛋白质富集的通路中涉及的代表性基因。(f) WT 和NAT之间活性差异的激酶。
03
基因组改变及其对转录组、蛋白质组和磷酸化蛋白质组的影响
全外显子组测序在 WT 中鉴定出 7 个显著突变基因,包括CTNNB1、WT1、AMER1、BRAF、TP53、CCNE1和PIK3CB。值得注意的是,在 TARGET WT 队列中也检测到了TP53、WT1、CTNNB1和AMER1的突变(图 3a)。几个基因是以前已知的致癌基因或抑癌基因,其中CTNNB1、AMER1和WT1与 WT 28相关。与 TARGET 数据库相比,本研究的队列具有相对较高的WT1和CTNNB1突变率,但TP53的突变率明显较低(图3a)。
将突变映射到癌症驱动通路上表明, Wnt/β-catenin 通路是 WT 中最常见的突变通路,并在细胞增殖以及胚胎肾脏发育中起关键作用。Wnt /β-catenin 通路相关突变样本(CTNNB1、AMER1突变)的表达谱显示 Wnt/β-catenin 通路发生激活,包括MYC、WNT5A、FZD1、NKD2和PLCB3在mRNA 水平上的上调(图 3b)。 Wnt/β-catenin通路突变样本差异表达基因富集分析发现,肌肉收缩通路、粘着斑等几种基质相关通路在突变样本中表达高于WT和NAT样本。此外,与肌肉功能、细胞结构和基质成分相关的Wnt/β-catenin通路下游基因,包括MYH3、MYL1和VIM,在突变样本中均高于WT样本和NAT样本(图 3b)。相应地,使用转录组数据分析Wnt/β-catenin突变样本的基质评分较高(图 3c)。这些结果表明肿瘤细胞的间质表型可能是通过Wnt/β-catenin通路激活来维持的。
拷贝数变异 (CNA) 分析发现了一些已知的 CNA,例如 1p/16q 的杂合性缺失 (LOH) 和 1q 的扩增。此外,他们还在 2p11.1、2q11.1、4p11、6p12.1、6q16.3、9q21.11、10q11.21、12q12、12q21.2、18q11.1 和 19q11 发现了几个显著的拷贝数扩增,并在 4q13.3 和 9q13 发现了拷贝数缺失(图 3d)。值得注意的是,在 TARGET-WT 队列中发现了 1q、4p、12q 和 19q 处的扩增,以及 1p、9q 和 16q 处的缺失。
接下来,他们进行了CNA与mRNA/蛋白质表达的相关性分析,以评估CNA对WT中mRNA和蛋白质表达的影响。结果显示,CNA与mRNA的相关性高于CNA与蛋白质的相关性,这可能是由于蛋白质翻译调控的复杂性所致。可能同时影响mRNA和蛋白质表达水平的CNA热点主要位于染色体6p、12p和12q区域(图 3e)。 CNA-mRNA 和 CNA-蛋白质相关性的联合分析鉴定出 225 个 CNA顺式调控基因,这些基因主要位于胞质带内,例如 1p、16q、12q、9q、6p、7p、7q、1q、13q、8p、6q 和 9p(图 3f),表明它们在肿瘤发生或进展中具有潜在的调控作用。此外,扩增的基因在与 RNA 剪接、调节 G0 到 G1 转换、DNA 复制、有丝分裂姐妹染色单体分离、染色质重塑和有丝分裂细胞周期的 G1/S 转换相关的通路中表现出高度富集。相反,主要位于 1p 和 16q 的缺失基因在电子传递链、羧酸分解代谢过程、脂肪酸氧化、己糖代谢过程和有机酸代谢过程等途径中富集(图 3g)。特别地,在位于顺式调控拷贝数扩增的基因中,大多数基因的表达水平与 EFS 和 OS 呈负相关。相反,顺式调控拷贝数缺失(1p 和 16q)中的基因的表达水平与无事件生存期(EFS)和总生存期(OS)呈正相关,进一步支持扩增基因具有促癌作用,而缺失基因具有抑癌作用。
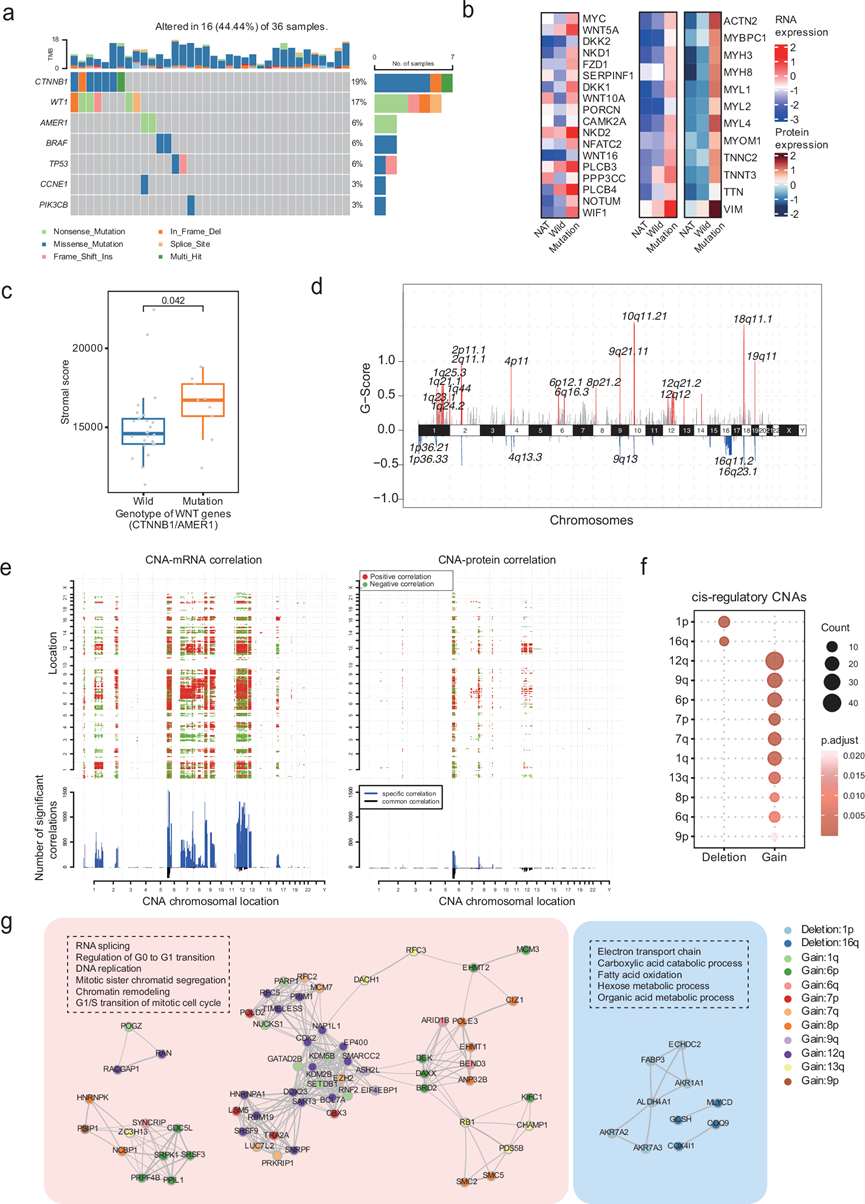
图3. 拷贝数改变和突变对 WT 中 mRNA 和蛋白质丰度的影响。
(a) 36个WT(Wilms肿瘤)样本中反复突变的基因。(b) Wnt/β-catenin通路和基质中相关基因的表达模式。(c) 基质评分的差异。(d) 频繁扩增和缺失的基因组区域。(e) 全基因组的 CNA-mRNA 和 CNA-蛋白质相关性。(f) 顺式调控 CNA 聚集在染色体胞质带上。(g) 具有潜在顺式调控效应的 扩增和缺失基因。
04
基于转录组和蛋白质组的WT分子分层
为了阐明WT肿瘤间的肿瘤间异质性,他们采用了一种整合方法,利用多组学数据对肿瘤样本进行分类。具体而言,将59个同时包含转录组和蛋白质组数据的WT样本分为三个不同的亚组,每个亚组均表现出独特的临床、病理和分子特征(图 4a)。
众所周知,WT 主要表现出三种主要病理类型(胚芽、上皮和间质组织学),并且胚芽组织学与较差的预后相关。值得注意的是,基于 HE 染色,亚组 1(S1)、亚组 2(S2)和亚组 3(S3)分别与胚芽显性、间质显性、上皮显性样本相关,表明病理分类与分子亚组之间具有高度一致性(图 4b)。这些结果表明 WT 的病理细胞类型是决定其分子特征的关键因素,而分子亚组比 HE 染色结果揭示的信息更复杂和准确。值得注意的是,S1 具有更高比例的高危患者和间变性病理。此外,S2 具有最高的基质评分,而 S3 表现出最高的免疫评分(图 4c),这表明 S2 和 S3 中不同的肿瘤微环境可能影响肿瘤生长动态以及肿瘤与周围环境之间的相互作用。
为了进一步表征WT的三个亚组,他们分析了每个亚组中差异表达的基因和通路。S1的特点是参与细胞周期和表观遗传调控以及DNA损伤反应(DDR)通路的mRNA和蛋白质表达增高,涵盖CDK1/2/4、EHMT1/2和HDAC2等基因。S2表现出在Wnt信号转导、肌生成、胶原和粘着斑通路中富集的蛋白质表达最高,例如COL1A1、COL3A1和MYH2。S3表现出免疫细胞标志物、主要组织相容性复合体和干扰素诱导蛋白表达增高,其中重要的蛋白质例如CD19、HLA-E和IFIT1(图 4d-e)。
磷酸化蛋白质组整合揭示了不同亚组间激酶活性和底物的差异,其中CDK1、CDK2、CDK5、CSNK1A1、MAPK8、GSK3A、PDPK1和GSK3B在S1中活性较高,而AKT1、RPS6KA1、CDK6和MAPK1在S3中活性较高,这凸显了各亚组内信号通路的异质性。值得注意的是,S1显示出不同的激酶活性热点,可能需要进行激酶靶向治疗(图 4f-g)。
有趣的是,S2 的特点是突出的CTNNB1突变,以及基质特征和 ECM 通路激活,这表明 Wnt 信号激活可能促进 WT 中间充质表型的维持。关键 CNA,包括 1p、16q 的缺失以及 1q、6p 和 12q 的增加,在 S1 中更为普遍,与细胞周期、表观遗传调控和 tRNA 调控相关通路激活等 S1 特征相一致。此外,他们使用在每个亚组中特别高表达的基因作为特征基因。使用最近的模板预测 (NTP) 模型,将来自 TARGET 数据库的 WT 样本分为 3 个亚组。生存分析显示 S1 具有较低的 EFS 率(图 4h)。该结果与 S1 中观察到的高危组织学和激活的细胞周期和 DNA 损伤反应 (DDR) 通路相符。由于 TARGET 和本研究的队列之间病理类型分布的差异,他们在 TARGET 队列中分离了弥漫间变性 WT (DAWT) 和复发性良好组织学特征的 WT (FHWT)。在复发性 FHWT 和 DAWT 中,与 S2 和 S3 相比,S1 仍然与更差的预后相关。此外,他们纳入了另一个包含原发性 FHWT 样本的公共数据库 (GSE31403),结果显示 S1 的 III-V 期样本比例明显较高,进一步表明 S1 与不良预后相关(图 4i)。

图4. WT 的蛋白质组学和转录组学分层以及相应的分子和通路特征。
(a) 通过谱聚类确定的亚组及其临床相关性。(b) 胚芽、基质和上皮成分比例的条形图。(c) 基质评分(左)和免疫评分(右)的箱线图。(d) RNA、蛋白质和磷酸化蛋白质水平的蛋白质组亚群中改变通路的热图。(e) 在 S1(左)、 S2(中)和 S3(右)中高表达的改变途径的特征基因的 mRNA 和蛋白质表达水平。(f) S1、S2 和 S3 中活化激酶的分类。(g) 分析亚群特异性磷酸化位点和相应激酶的热图。(h) TARGET 队列中 WT 亚群的无事件生存期(左)和总生存期(右)的 Kaplan-Meier 图。(i) 数据库 GSE31403 中 S1、S2 和 S3 亚型中 I、II、III、IV 或 V 期肿瘤的比例。
05
WT肿瘤发生的肾脏发育视角
除了常见的致瘤分子特征外,他们还鉴定了与 WT 中肾脏发育相关的通路的激活,特别是那些参与胚胎发育和 Wnt 信号通路的通路(图 2c)。早期肾脏发育经历四个阶段:后肾间充质(MM)、输尿管芽(UB)、帽间充质(CM)和肾囊泡(RV)。多项研究表明,WT 表现出与早期肾脏发育相似的基因谱。例如,肾脏发育的关键调节因子,如SIX2、SALL1、WT1和EYA1,在 WT 中在 mRNA 和蛋白质水平上均上调。此外,他们还收集了代表 12 个肾脏发育阶段的 103 个特征基因,并使用基因集变异分析(GSVA)算法分析了它们在每个样本中的表达。S1 与 MM 和 CM 密切相关,S2 与系膜细胞和肾间质(RI)密切相关,S3 与 S 形体密切相关(图 5a)。这些发现与每个分子亚组内的主要病理类型相一致,表明不同 WT 肿瘤细胞群的潜在起源。WT 显示出反映不同发育阶段的不同基因特征,包括 MM、CM、RV、系膜细胞和 RI。值得注意的是,对应于UB、输尿管尖端和相对成熟的细胞类型(如足细胞、近端小管和远端小管)的特征缺失,这意味着WT肿瘤发生具有特定的发育轨迹。TARGET-WT队列显示出这些发育阶段的相似表达模式,进一步支持WT与早期胚胎肾脏发育之间的密切关联,特别是间充质-上皮转化(MET)之前的阶段,例如MM和CM。MET是早期肾脏发育的关键过程,由几个具有关键作用的基因控制。在本研究的队列中,他们观察到蛋白质水平上关键 MET 促进基因(CDH6、CDH4和FGF1)的下调,这表明在 WT 肿瘤发生中 MET 过程可能存在失调(图 5b)。
对不同亚组的关键转录因子 (TF) 进行分析表明,与 NAT 相比,与 CM 和 RV 相关的 TF 在 WT 中高表达,尤其是在 S1 中(图 5c)。此外,对早期胚胎肾脏发育至关重要的 TF,例如SOX11、SALL1、WT1和MAZ,在 WT 样本中显著过表达。这些TF 与 WT 中各自的靶基因表达表现出很强的相关性,突出了它们在 WT 发育中的潜在调控作用(图 5d)。
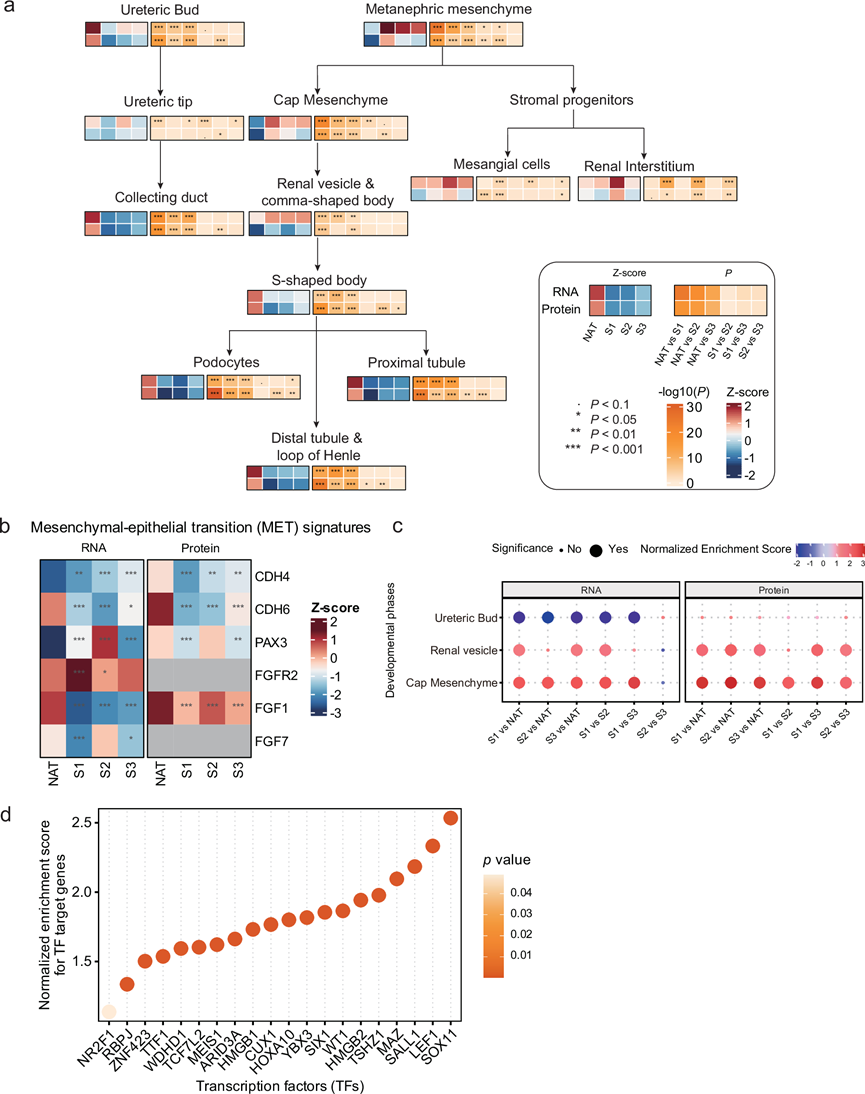
图5. WT的肾脏发育相关特征及分子亚群的差异。
(a) 通过 ssGSEA 算法对肾脏的 12 个发育阶段进行的评分。(b) NAT、S1、S2 和 S3 WT 样本中间充质-上皮转化特征基因在 mRNA 和蛋白质水平上的表达。(c) NAT、S1、S2 和 S3 中肾脏发育阶段相关基因集的归一化富集评分 (NES)。(d) WT样本中TF靶基因的归一化富集得分。
06
基于整合蛋白质组学数据的WT免疫景观
为了表征WT的免疫微环境,他们采用 CIBERSORTx 方法,基于基因表达数据估算肿瘤组织内免疫细胞的相对丰度。通过将本组队列与 TCGA 数据库中的成人肾癌样本和 TARGET 数据库中的 WT 样本进行比较,发现 WT 样本的免疫细胞比例显著低于成人肾癌,例如肾透明细胞癌 (KIRC) 和肾乳头状细胞癌 (KIRP)(图 6a)。此外,他们观察到免疫细胞比例与 TMB 之间无相关性(图 6b)。
为了进一步分析WT的免疫微环境,他们利用 CIBERSORTx 方法估算了 22 种不同免疫细胞类型的相对丰度。WT 中大多数免疫细胞的丰度始终相对较低,包括 B 细胞、细胞毒性细胞、巨噬细胞和抗原呈递机制,这表明与成人肿瘤相比,WT 具有更强的免疫抑制表型,类似于其他儿童胚胎肿瘤。值得注意的是,M2 巨噬细胞、γ-δ T 细胞、活化肥大细胞、CD8 T 细胞和静息 CD4 记忆 T 细胞在 S3 亚群中显著富集(图 6c)。这些发现表明 S3 具有更高的免疫细胞浸润,可能代表免疫富集的亚型,这与不同亚组中 CD4、CD8、CD45 和 PD-L1 的免疫组织化学 (IHC) 结果一致。尽管与成人肾肿瘤相比,WT 中观察到的总体免疫浸润较低,但 S3 表现出更高的免疫细胞比例和增加的免疫细胞丰富度,特别是抗原呈递机制和细胞毒性细胞。此外,S3 显示出比 S1 和 S2 亚组更高的趋化因子、细胞因子和干扰素表达水平(图 6d)。除了免疫浸润之外,免疫抑制分子在塑造肿瘤的免疫表型和影响其对免疫治疗的反应性方面起着至关重要的作用。本研究的研究结果表明S3可能存在免疫逃避机制,其证据是免疫检查点基因(包括CD274和BTN3A1)在mRNA和蛋白质水平上的表达显著升高(图 6e)。这些结果凸显了S3的免疫调节特性,提示其可能成为免疫治疗的有希望的候选药物。
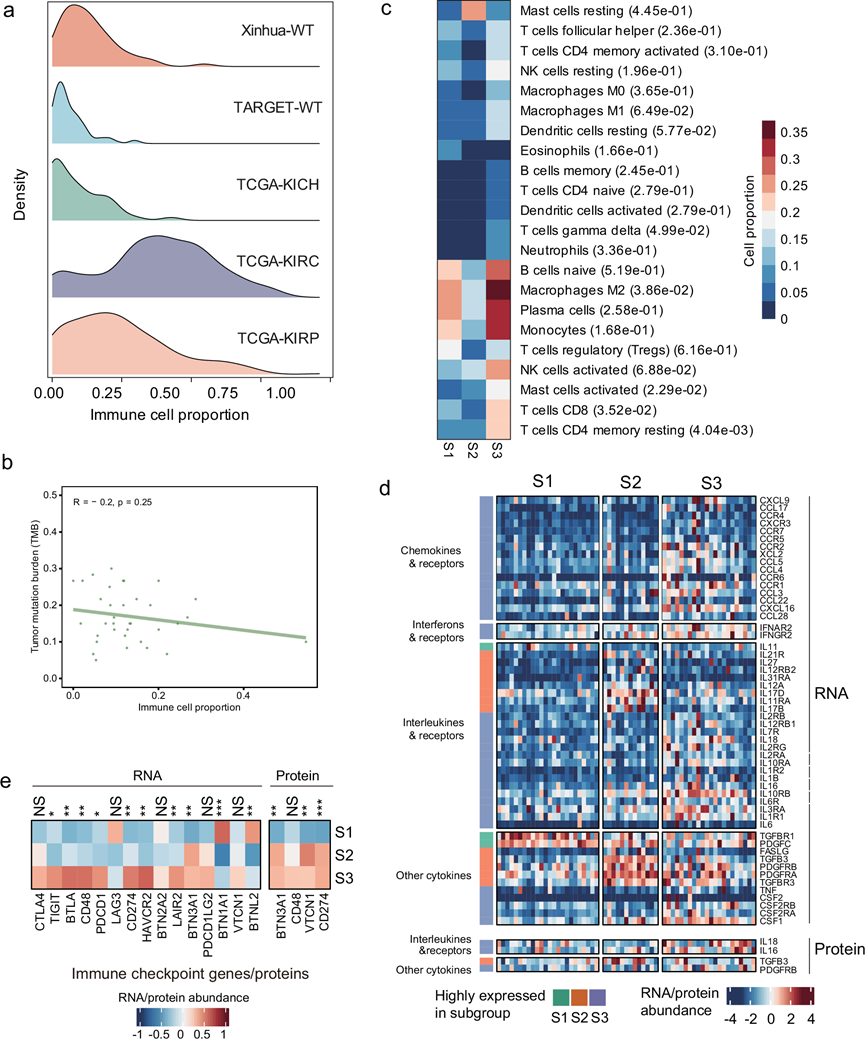
图6. WT 在 mRNA 和蛋白质水平上的免疫特征。
(a) 免疫特征评分的岭图。(b) 肿瘤突变负荷 (TMB) 与免疫特征评分之间的相关性分析。(c) 免疫细胞的细胞丰度。(d) S1、S2 和 S3 中趋化因子、干扰素、白细胞介素和其他细胞因子的RNA(上)和蛋白质(下)丰度。(e) S1、S2 和 S3 中免疫检查点基因/蛋白质的RNA(左)和蛋白质(右)丰度。
07
通过蛋白质组学分析确定治疗策略
精准医疗通过选择性靶向致癌通路(包括突变、CNA、差异表达蛋白和激酶)在癌症治疗中发挥着至关重要的作用。通过综合分析,他们共确定了 39 个潜在治疗靶点(图 7a)。值得注意的是,其中一些候选基因与 WT 中的不良预后显著相关,并且可以成为 FDA 批准药物的靶点。这些基因在促肿瘤通路中富集,例如细胞周期、TP53 活性调节和 Wnt 信号通路(图 7b)。在这些潜在的药物靶点中,CDK2-RB1-E2F 轴已成为一个经过充分研究的通路,与驱动各种恶性肿瘤中的细胞增殖有关。在WT中,该轴的激活受到CDK2表达和激酶活性升高、RB1过度磷酸化以及E2F靶基因上调(尤其是在S1中)的支持(图 7c)。此外,在WT细胞系中使用小分子抑制剂BIX-01294(S8006,Selleck)抑制CDK2,可导致RB1磷酸化水平降低,从而证实了CDK2对WT细胞中RB1磷酸化的调控作用。
EHMT2 是一种组蛋白赖氨酸 N-甲基转移酶,通过单甲基化和二甲基化调控组蛋白 H3,发挥转录抑制因子的作用,与肝细胞癌和黑色素瘤相关。值得注意的是,EHMT2 在 S1 组 mRNA 和蛋白质水平上均高表达(图 7a),且其高表达与 TARGET 队列中 EFS 和 OS 缩短相关。此外,在数据库 GSE31403 中,与 I 期 WT 样本相比,EHMT2 在 II、III、IV 和 V 期中高表达。鉴于 EHMT2 的功能意义,他们进行了体外实验来研究其功能作用和潜在机制。首先,通过对 18 组 WT 和 NAT 样本进行 Western blot 分析,证实了 WT 中 EHMT2 的高表达(图 7d)。其次,在 WT 细胞中使用 EHMT2 特异性小干扰 RNA(siRNA)敲低 EHMT2 导致 H3K9 二甲基化水平下调(图 7e)。功能测定表明,EHMT2 沉默诱导 G1 阻滞并抑制细胞增殖。此外,qPCR 分析表明,EHMT2 沉默导致 WT 细胞中 pre-rRNA 水平降低,表明 EHMT2 参与了 rRNA 调控(图 7f)。此外,他们对敲低或未敲低EHMT2的WT细胞进行了转录组分析(RNA-seq),以评估EHMT2对WT细胞中基因表达的影响。结果显示,参与Wnt信号转导、细胞周期调控和肾发生等基因显著下调,而上调的基因与自噬、凋亡和程序性细胞死亡相关(图 7g)。具体而言,在EHMT2抑制后,细胞周期、Wnt/β-catenin信号转导和肾发生等关键调控因子显著下调(图 7h),这表明EHMT2可能通过这些途径调控细胞增殖和细胞周期进程。
为了进一步阐明EHMT2在WT细胞中的潜在治疗靶点,他们用不同剂量的EHMT2小分子抑制剂BIX-01294处理WT细胞系24小时。EHMT2的主要甲基化靶点H3K9二甲基化显著降低,而组蛋白H3总蛋白水平保持不变(图7i)。这些结果表明EHMT2在WT细胞H3K9二甲基化中发挥着关键作用,并突显了EHMT2作为WT细胞有前景的生物标志物和潜在药物靶点的潜力。
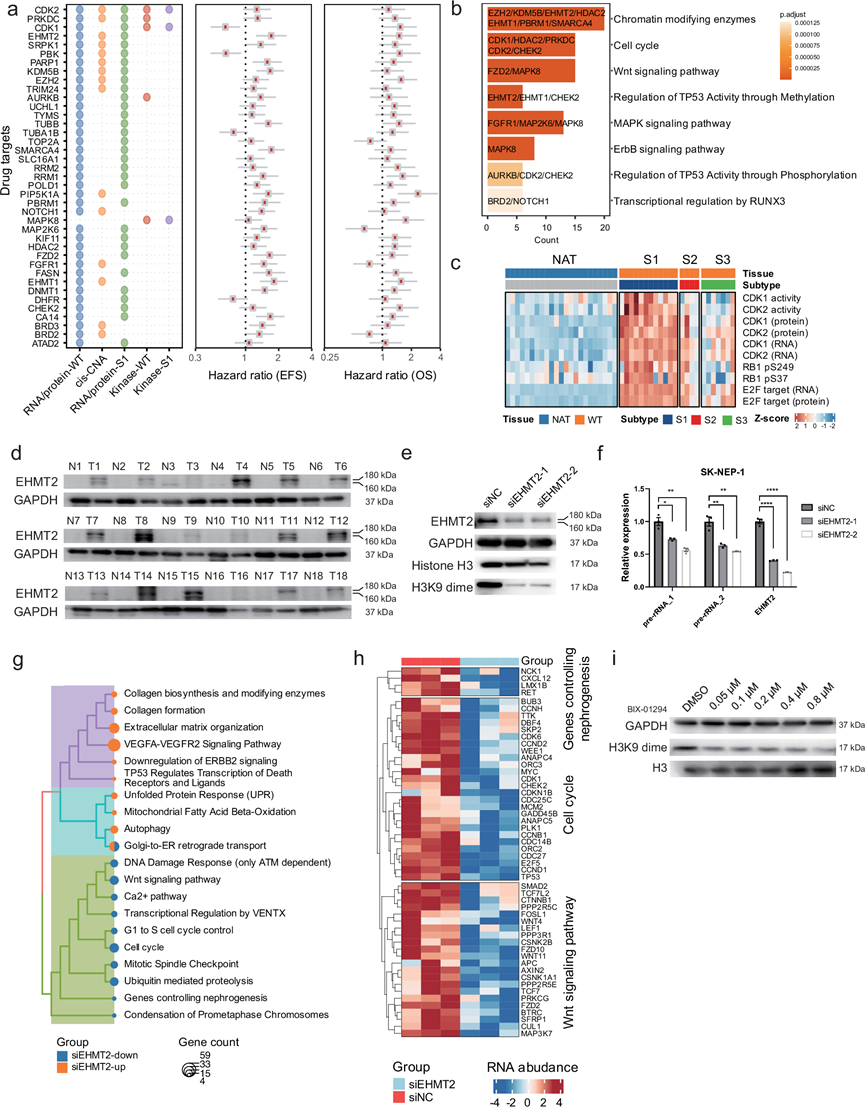
图7. 预后生物标志物和潜在治疗靶点的识别和验证。
(a) 从多组学数据分析得出的 FDA 批准药物的潜在药物靶点(左)及其使用 TARGET-WT 队列(n = 125)的 EFS 和 OS 的风险比(右)和 95% 置信区间(CI)。(b) 潜在药物靶点富集的通路。(c) 热图描绘了 CDK1 和 CDK2 的激酶活性和 RNA/蛋白质表达水平、RB1(pS249 和 pS37)的磷酸化水平以及 E2F 靶基因的 RNA 和蛋白质表达水平。(d-e) 免疫印迹分析。(f) SK -NEP-1 细胞中 EHMT2 敲低后 pre-rRNA 的 mRNA 表达。(g) EHMT2 敲低后下调和上调基因的通路富集结果。(h) 与控制肾脏发生、细胞周期和 Wnt 信号通路相关的差异表达基因 RNA 丰度热图。(i) 免疫印迹分析。
+ + + + + + + + + + +
结 论
本研究对 WT 及其肿瘤旁正常肾脏组织进行了蛋白质组学、磷酸化蛋白质组学、转录组学和全外显子组测序整合分析。多组学方法揭示了预后基因变异、独特的分子亚群、免疫微环境特征以及潜在的生物标志物和治疗靶点。基于蛋白质组和转录组的分层分析识别出三个具有独特特征的分子亚群,它们与胚胎肾脏发育不同阶段的不同组织病理学亚型和推测的细胞来源相关。值得注意的是,本研究发现 EHMT2 是一个有希望的预后生物标志物和治疗靶点,与表观遗传调控和 Wnt/β-catenin 通路相关。在这项工作中,本研究对 WT 进行了全面的分子表征,为其发病机制提供了宝贵的见解,并为未来的治疗发展提供了基础资源。
+ + + + +



