

 English
English文献解读|Nat Commun(15.7):多组学分析揭示了烟草中代谢物生物合成的调控网络
✦ +
+
论文ID
原名:Multi-omics analyses reveal regulatory networks underpinning metabolite biosynthesis in Nicotiana tabacum
译名:多组学分析揭示了烟草中代谢物生物合成的调控网络
期刊:Nature Communications
影响因子:15.7
发表时间:2025.11.24
DOI号:10.1038/s41467-025-65299-6
背 景
烟草是一种重要的工业作物,是植物科学的模式生物,也是生产蛋白质和小分子化合物的理想物种。然而,目前对烟草在自然田间栽培条件下的系统生物学研究仍然匮乏。
实验设计
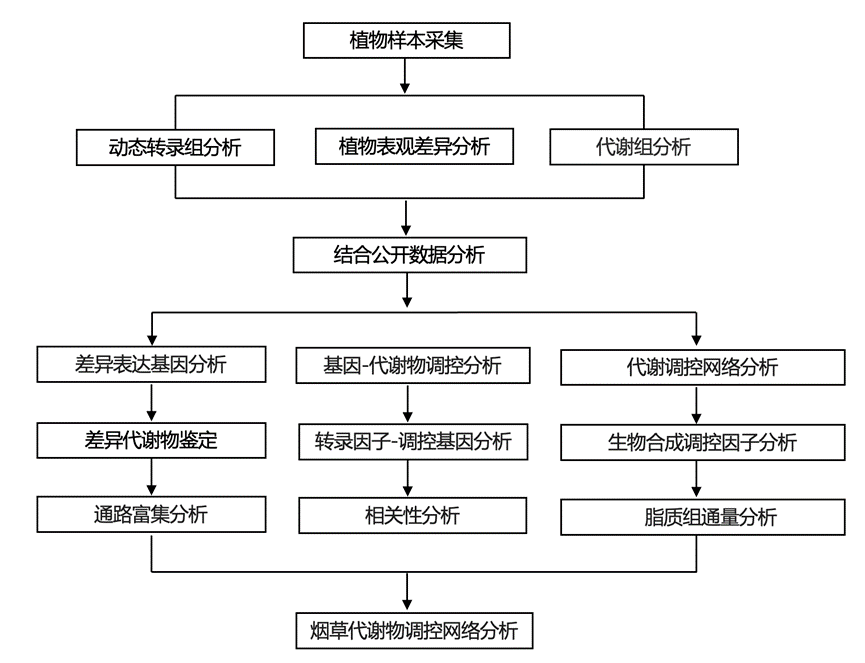
结 果
01

环境影响烟草叶片的发育、基因表达和代谢物积累模式
K326是全球广泛种植的烟草模式品种,分析其代谢调控网络具有重要的代表性。此外,考虑到该烟草将用于田间工业化生产,研究团队在两个环境差异显著的典型烟草种植区[高海拔山区(HM)和低海拔平原(LF)]开展了田间种植试验。为了解不同环境因素对烟草生长发育的影响,他们分析了烟草采样期间两个区域的气候数据。在采样期间,LF和HM的净太阳辐射、相对湿度和降水量均无显著差异。两地间的主要气候差异体现在温度方面,包括降雨量、温差和最高温度(图 1a-f)。LF和HM处理组的温度范围、平均温度和最高温度均存在显著差异,但最低温度无显著差异(图 1d-f)。既往研究表明,温度是影响烟草生长和代谢稳态的关键因素。因此,温度变化引起的基因表达和代谢物积累的扰动可以提高后续代谢调控网络的准确性和稳定性。
为了建立标准化的烟叶发育体系,他们采用了摘心策略(图 1g)。摘心严格基于LF和HM植株相同的发育阶段。因此,虽然不同地点间的生态差异导致了生长速率、开花时间和成熟期的差异),但在同一发育阶段同步摘心最大限度地消除了先前存在的发育差异。在开花当天,人工去除主茎顶端的花序以及一些新叶,确保每株烟叶保留18片健康叶片。摘心消除了顶端优势,停止了生殖生长,并将更多养分用于次生代谢物的合成,从而使烟叶的营养系统标准化。摘心后,上部叶片仍在生长,而下部叶片更容易受到病虫害侵袭,光照减少,并已进入衰老过程。因此,他们选择中间位置的第8和第9片叶子进行取样(图 1g)。此外,在摘心后第20天开始取样,以避开摘心后烟草植株内激素水平紊乱、酚类、生物碱、色素和萜烯类等各种代谢物水平显著波动的时期,因为这些波动可能会干扰研究。
为了解代谢动态及其相关机制,他们采集了LF和HM烟草在摘心后20至40天的叶片,并进行了转录组学分析(RNA-seq)和代谢组学分析。在成熟过程中,HM和LF的叶片颜色存在差异(图 1h)。因此,他们检测了叶片中的叶绿素含量。结果表明,LF叶片的比叶重(SLW)标准化SPAD值显著高于HM叶片,提示LF叶片的衰老速度慢于HM(图1i)。共鉴定并定量了所有样本中的633种代谢物,并根据其代谢特征将其分为14个主要类别。层次聚类分析显示,代谢物的位置特异性高于时间进程特异性。主成分分析(PCA)也表明,代谢物均可按位置分为两组(图 1j)。此外,在LF和HM中,20至40 DAT(摘心后天数)期间均可根据时间进程分为两个聚类,LF的聚类转换点为28 DAT,HM的聚类转换点为30 DAT。
为了探究烟叶发育的遗传基础,他们收集了与代谢组学样本同批次收获的烟叶,每个样本重复三次进行RNA-seq。RNA-seq重复结果显示出高度正相关性。在22个样本中,共检测到34839个蛋白质编码基因表达。层次聚类分析和主成分分析(PCA)表明,位点特异性大于时间特异性,提示位点在基因表达中起着重要作用(图 1k)。来自两个位点的样本在不同时间点也能分别聚类。总体而言,在低LF和HM的烟叶发育过程中,代谢组和转录组均表现出显著的发育特异性。这些结果表明,环境对代谢物积累和基因表达有很强的影响,而代谢物的积累模式在成熟过程中会随时间逐渐改变。
为了解代谢物和基因表达随时间的变化,他们对LF和HM中的差异基因表达和代谢物生成进行了时间序列分析。在LF中共鉴定出14595个差异表达基因(DEG)和308个差异丰度代谢物,而在HM中鉴定出6943个DEG和196个差异丰度代谢物。LF和HM之间共有3476个时间序列DEG和134个时间序列差异丰度代谢物。KEGG和GO富集分析突显了LF和HM之间时间序列DEG功能上的显著差异(图 1l)。例如,GO富集分析还揭示了这些基因在HM中具有更多样化的生物学功能。 KEGG分析表明,HM中特定时间基因的功能富集于光合作用、类胡萝卜素生物合成和游离脂肪酸降解等过程(图 1l)。对特定时间差异表达代谢物的KEGG富集分析也表明,LF和HM之间代谢物的生物学功能存在显著差异(图1m)。
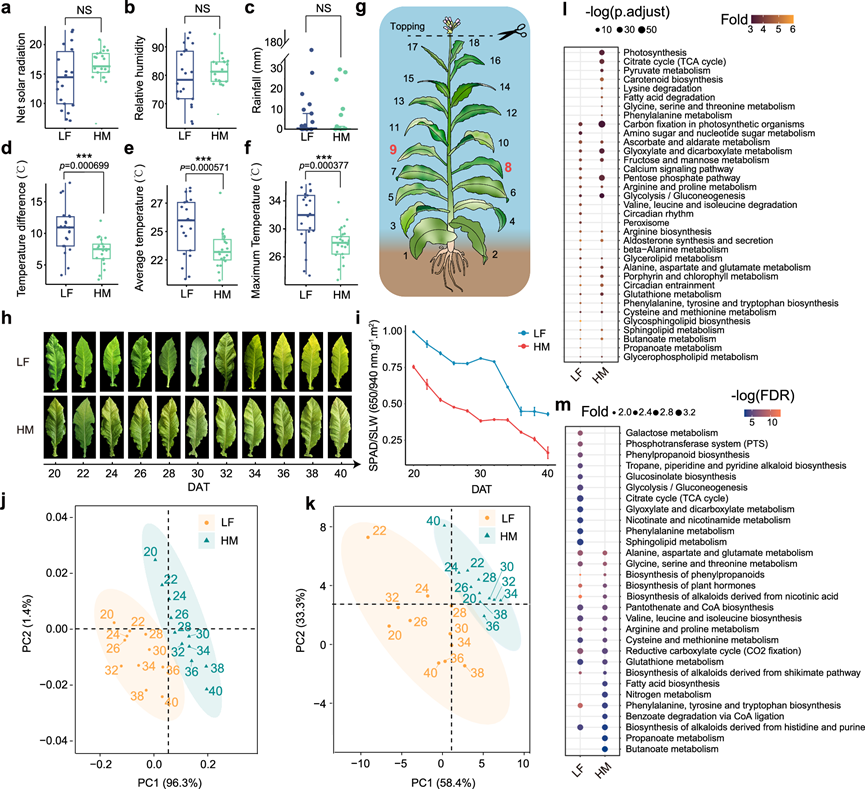
图1. 实验设计示意图及代谢组和转录组分析总结。
(a-f) 贵阳(LF)和龙山(HM)在摘心后20至40天(DAT)的净太阳辐射量、相对湿度、降雨量、温差、平均温度和最高气温的比较。(g) 烟草取样示意图。(h) LF和HM在摘心后20至40天的烟草中部叶片。(i) LF和HM在摘心后20至40天的烟草中部叶片的土壤植物分析发育(SPAD)/比叶重(SLW)比值。(j-k) 22 个烟草样本的代谢组和转录组数据的 PCA 结果。(l-m) 差异表达基因和代谢物的 KEGG 富集分析结果。
02
多聚类分析能够更清晰地描绘复杂的基因-代谢物调控图谱
调控基因及其靶标通常在较长的时间尺度和不同的组织中保持相似的表达模式。基于这些原理,我们融合了多种聚类方法,包括加权基因共表达网络分析(WGCNA)、基于Pearson相关系数(PCC)的共调控分析和k均值聚类,以评估基因与代谢物之间的关联,并采用基尼系数结合机器学习方法来捕捉非线性关系(图 2a)。首先,排除了与任何代谢物均无相关性的基因。最终共鉴定出25984个基因。随后,他们将这些基因和代谢物分为9个k均值聚类(图 2b)。为了校准聚类内基因-代谢物调控关系,他们采用了一种直接基于PCC的共调控方法(图 2b)。通过分析PCC与k均值聚类结果之间的映射关系,发现不同的聚类方法在聚合调控对方面存在偏差,包括一些已知的代谢相关基因。通过融合多种聚类方法,提高了二维调控关系的准确性,并获得了最终的聚类结果,涵盖了630个代谢物和21527个基因(图 2c)。这些聚类在烟草叶片发育过程中呈现出一致且独特的表达模式。例如,聚类2中基因和代谢物的表达持续增加,而聚类3中基因和代谢物的表达持续降低,表明烟草基因的丰度模式与主要代谢途径密切相关(图 2c)。
为了更精确地鉴定调控基因,他们收集了所有烟草转录因子(TF)的信息。在转录组水平上,共检测到2390个TF作为调控因子。此外,还鉴定出22569个结构基因(SG)作为潜在靶标。通过对每个聚类中TF家族的富集分析,发现每个聚类都显著富集了特定的TF类别,表明它们可能在调控同一聚类中相应的SG和代谢物(ME)方面发挥关键作用。多簇分析能够更清晰地展现复杂的全基因组调控关系,为构建全基因组代谢调控网络(GMRN)奠定了基础(图 2c)。
为了捕捉非线性调控关系并增强全局调控网络的全面性,他们计算了每个SG、TF和ME之间的GINI系数,并采用简单的随机森林方法对调控重要性进行排序,从而有助于筛选候选调控基因或代谢物。在24959个基因和633个代谢物中,共鉴定出2332526个高置信度GINI调控对,并计算了每个SG、TF和ME的重要性排序。接下来,我们分析了这些基因的聚类和调控情况。同一聚类内的个体表现出比随机配对更强的相关性或更高的GINI系数(图 2d)。PCC和GINI调控对之间的重叠显著高于随机对照组,表明调控关系挖掘的准确性和稳健性(图 2e)。在所有聚类中,ME-ME、ME-SG 和 ME-TF 对的调控关系均不平衡(图 2f)。ME-ME、ME-SG 和 ME-TF 对的 PCC 和 GINI 系数分布也从统计学上证实了这一结果(图 2g)。为了验证数据的准确性,他们从文献中收集了已知调控不同代谢途径(包括脂质、糖类、苯丙烷和萜烯途径)的TF的数据。结果表明,这些 TF 及其靶标聚集在一起,且它们的 PCC 和 GINI 系数均高于平均值,表明基因-代谢物相关性聚类的准确性(图 2h)。由于 ME-ME、ME-SG 和 ME-TF 之间的调控关系不平衡,我们采用逐步分析方法,通过 ME-SG-TF 级联来鉴定 CGA 和 DHA 的调控因子。与其他SG相比,NtLOX2与DHA的相关性最高(图 2i),而Nt4CL2A(编码肉桂酸4-羟化酶)和NtPAL2(编码苯丙氨酸解氨酶)与CGA的相关性最高(图 2k)。类似地,与其他TF相比,TEOSINTE BRANCHED 1/CYCLOIDEA/PCF (TCP)转录因子NtCYC(CYCLOIDEA)与NtLOX2的相关性最强(图 2j),而MYB转录因子NtMYB28与Nt4CL2A和NtPAL2的相关性最强(图 2l)。随后,他们计算了NtCYC与DHA以及NtMYB28与CGA的GINI系数和重要性排序。与PCC结果一致,GINI系数和重要性排序如下:NtLOX2-DHA、NtCYC-NtLOX2、Nt4CL2A /NtPAL2-CGA和NtMYB28-Nt4CL2A/NtPAL2之间的关联显著高于其他涉及 SG 或 TF 的调控关系(图 2m-p)。这些结果表明,NtCYC和NtMYB28可能分别是 DHA 和 CGA 代谢的关键调控因子。
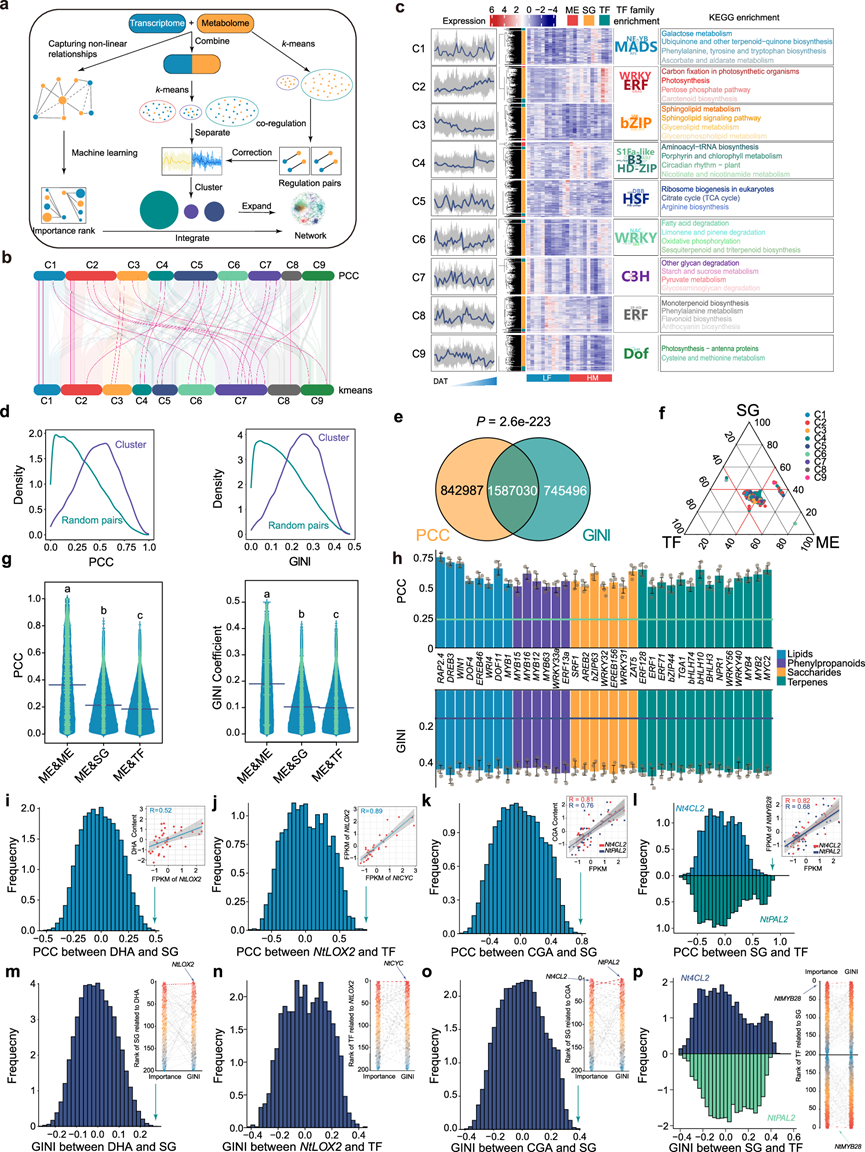
图2. 代谢物和基因的多聚类分析。
(a) 基因-代谢物聚类分析的工作流程。(b) 两种方法间不同聚类结果的映射关系。(c) 最终聚类的特征。(d) 同一聚类(紫色)与随机(绿色)基因-基因或基因-代谢物对之间的Pearson相关系数 (PCC) 和基尼系数。(e) 通过不同方法识别出的调控对的显著重叠表明了调控关系的稳健性。(f) 三元图显示了代谢物 (ME) 与结构基因 (SG)、转录因子 (TF) 与 ME 之间相关性分布的偏差。(g) 代谢物之间的 PCC 和基尼系数显著高于代谢物与 SG 或 TF 之间的 PCC 和基尼系数。(h) 已知代谢相关转录因子(TF)及其靶基因在同一基因聚类中的表达值之间的PCC和GINI系数。(i) DHA与SG表达水平之间的PCC相关性。(j) NtLOX2与TF表达水平之间的PCC相关性。(k) CGA与SG表达水平之间的PCC相关性。(l) Nt4CL2A、NtPAL2与 TF表达水平之间的 PCC 相关性。(m) DHA 与 SG 表达水平之间的 GINI 相关性。(n) NtLOX2与TF 表达水平之间的 GINI 相关性。(o) CGA 与 SG 表达水平之间的 GINI 相关性。(p) Nt4CL2A、NtPAL2与 TF 表达水平之间的 GINI 相关性。
03
构建全基因组代谢调控网络并进行表征
为了构建烟草叶片发育过程中的代谢调控网络,他们扩展了二维调控关系,包括通过多次聚类获得的PCC和GINI调控对,并重复分析了五次(分别使用三个独立的转录组或代谢组重复样本、平均表达水平以及PCC-GINI合并的调控对)。随后,将结果合并,得到最终网络,该网络包含3175516个高置信度的调控对(图 3a)。为了精确表征烟草的代谢调控网络(GMRN),利用NetworkX软件,采用传递性、平均特征向量中心性和平均路径长度等网络参数来定量GMRN的拓扑结构。烟草GMRN表现出较高的传递性(0.101)、平均特征向量中心性(0.688)和平均路径长度(2.638)。他们将2802个仅表现出基于基尼系数的调控关系的基因分配到暂定的聚类0中,以便分析它们与其他聚类的跨聚类相互作用。GMRN中不同聚类之间的映射关系表明,聚类内调控对占主导地位,但跨聚类调控关系仍然存在,尤其是在聚类0中(图 3a)。枢纽基因和代谢物在五个网络中的分布一致(图 3b-c)。关键基因的富集分析表明它们参与多种代谢途径,可能代表生物体发育中的重要代谢过程(图 3d-e)。此外,关键基因中排名前三的转录因子家族是乙烯反应因子(ERF)、WRKY和MYB家族,这些家族因其在代谢调控中的积极作用而闻名(图 3f)。
鉴于ERF和WRKY转录因子在植物代谢调控中潜在的关键作用以及在枢纽基因分析中的高排名,他们提取了以ERF或WRKY转录因子为中心的代谢调控网络,与GMRN相比,这些网络的拓扑结构发生了变化。与GMRN相比,ERF-GMRN表现出更高的传递性(0.637)、更低的平均特征向量中心性(0.0523)和更短的平均路径长度(2.368)。ERF-GMRN包含2442个节点和57798个调控对(图 3g)。在网络中鉴定出连接度最高的五个ERF转录因子,包括ERF1、ERF49、ERF167、脱水响应元件结合蛋白3 ( DREB3 ) 和与APETALA2.4相关的蛋白 (RAP2.4)(图 3g)。ERF1和RAP2.4此前已被证实能够调控蜡质合成。DREB3参与ABA信号通路和抗旱性,并能调控磷脂和碳水化合物代谢。ERF49与植物对高温和干旱胁迫的响应相关,而ERF167的功能尚不清楚。GMRN揭示了ERF1和DREB3之间一些已知的调控关系,包括那些已通过实验室实验验证的关系。ERF1主要与多种脂质代谢的潜在级联调控相关,而DREB3则与磷脂和碳水化合物代谢相关(图 3h)。他们随机选取了基于PCC或GINI系数鉴定的ERF1和DREB3靶基因,并进行了双荧光素酶报告基因检测。结果表明,ERF1能够激活脂质转运蛋白生物素羧基载体蛋白1(BCCP1)和过氧化物酶基因过氧化物酶1(POD1)的表达,而单糖转运蛋白单糖转运蛋白6(MST6)和热休克转录因子A-2(HSFA2)则受到DREB3的正向调控(图 3i)。
与GMRN相比,WRKY-GMRN表现出更高的传递性(0.499)和更长的平均路径长度(3.295),但平均特征向量中心性更低(0.0253)。WRKY-GMRN中连接性排名前三的转录因子是WRKY42、WRKY22和WRKY11(图 3j)。WRKY42调控叶绿素降解和类胡萝卜素合成。WRKY11受WRKY22调控,促进花青素、类黄酮和木质素的合成,并能增强植物对病原体的抗性。GMRN恢复了WRKY转录因子的一些靶标(图 3k)。我们随机选择了一些PCC或GINI调控对进行验证。结果表明,WRKY11促进黄烷醇合酶基因Flavanol Synthase (FLS)和半乳糖醛酸转移酶基因Galacturonosyltransferase-Like 9 (GATL9)的表达,而WRKY42正向调控番茄红素β-环化酶基因Lycopene β-Cyclase (LCYB)和谷氨酰胺合成酶基因Glutamine 1 (Glu1)的表达(图 3l)。这些结果不仅进一步验证了GMRN的准确性,而且有助于重新构建烟草中ERF和WRKY转录因子的调控网络。总而言之,GMRN将有助于代谢调控基因的发现。
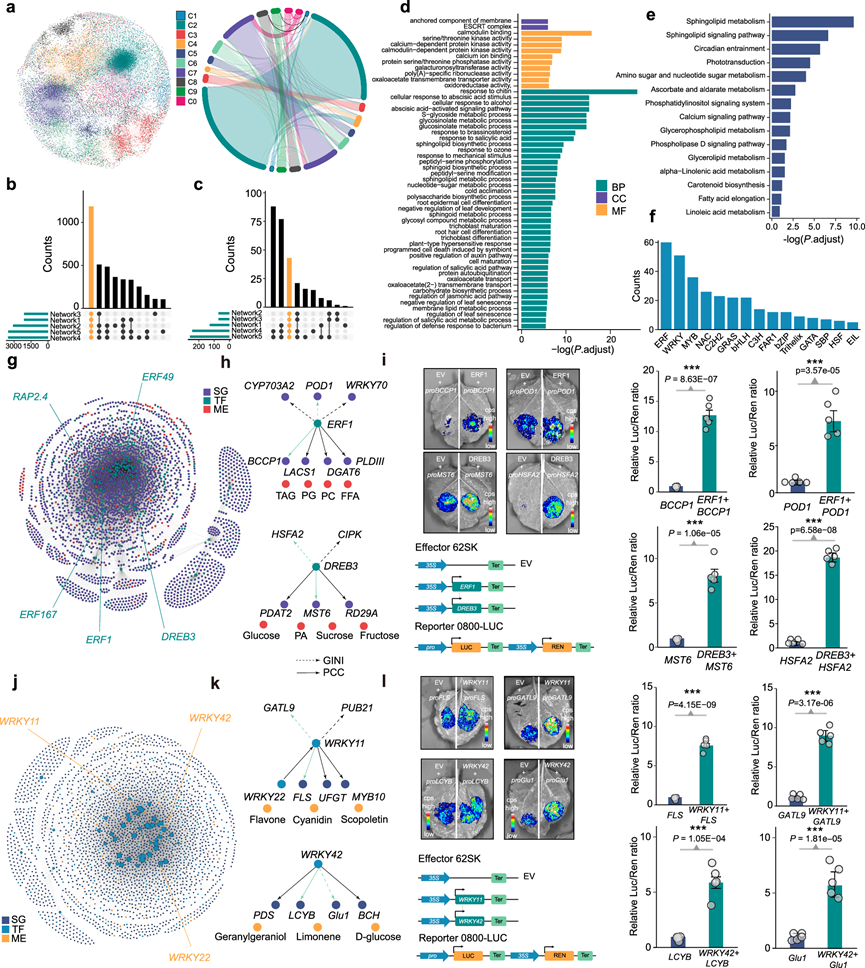
图3. GMRN 提供了关于转录因子对代谢物调控的见解。
(a) 涵盖9个共响应聚类和1个GINI特异性聚类(C0)的全基因组GMRN图谱(左图)以及GMRN中不同聚类间调控对的映射关系(右图)。(b-c) 来自不同数据源的GMRN之间重叠的关键基因和代谢物的数量。(d-e) GMRN中枢纽基因的GO和KEGG富集分析。(f) GMRN预测的关键调控因子总数最多的前15个TF家族。(g) 以ERF基因为中心的子网络。(h) GMRN 回收了与不同代谢物相关的ERF1和DREB3的靶基因。(i) ERF1和DREB3靶基因调控的验证。(j) 以WRKY 基因为中心的亚 GMRN。(k) GMRN回收了与不同代谢物相关的WRKY11、WRKY22和WRKY42的靶基因。(l) 验证WRKY11和WRKY42靶基因的调控作用。(m) KEGG通路富集分析。
04
GMRN有助于鉴定参与羟基肉桂酸生物合成的关键调控因子
接下来,他们利用GMRN鉴定了调控靶代谢物生物合成的关键转录因子。聚类分析结果表明,NtMYB28可能通过调控Nt4CL2A和NtPAL2的表达来调控CGA的生物合成。苯丙氨酸解氨酶和肉桂酸4-羟化酶是CGA生物合成途径中的限速酶。为了解NtMYB28的调控关系,他们从GMRN中提取了一个以靶代谢物为中心的亚调控网络(图 4a)。苯丙素生物合成基因及其相关转录因子的表达模式与羟基肉桂酸的积累模式高度一致(图 4b)。NtMYB28的 GMRN 中共同调控的基因和代谢物的 KEGG 富集分析表明,它们的分子功能主要富集于苯丙氨酸代谢和苯丙素生物合成。
转录激活活性和亚细胞定位分析表明,NtMYB28具有TF的功能(图 4c)。为了进一步研究NtMYB28是否直接诱导苯丙素生物合成基因的表达,他们克隆了Nt4CL2A和NtPAL2的启动子。Nt4CL2A和NtPAL2的启动子区域(ATG 上游 1000 bp 区域)包含 11 个 MYB 结合基序,包括 AC I-III、次级代谢响应元件 (SMRE)-I 和 II。 LUC 活性测定、酵母单杂交测定 (Y1H) 和电泳迁移率变动测定 (EMSA) 的综合结果(图4d-f)表明,NtMYB28 蛋白可以通过 AC-I 基序直接与Nt4CL2A和NtPAL2的启动子结合(图 4d-f)。
他们构建了两个独立的NtMYB28过表达株系(NtMYB28过表达株系(OE)#8和13)和两个独立的NtMYB28突变株系[NtMYB28敲除株系(KO)#11和14]。Nt4CL2A和NtPAL2的mRNA水平在OE株系中显著升高,而在NtMYB28-KO株系中则显著下调(图 4g-h)。与野生型(WT)烟草相比,两个NtMYB28-OE株系中羟基肉桂酸的含量增加了1.2-2.0倍,而NtMYB28-KO株系中羟基肉桂酸的含量则显著降低了1.5-2.3倍(图 4i-j)。主成分分析(PCA)显示,两个OE株系和两个KO株系的转录组分别与WT存在显著差异。这些DEG与GMRN中预测的NtMYB28靶基因的重叠度显著高于随机对照组(图 4k),表明GMRN能够准确预测NtMYB28的靶基因。用于构建GMRN中苯丙素生物合成途径亚调控网络的SG的表达水平在WT、OE和KO株系之间存在差异(图 4l)。此外,WT和OE株系间差异表达基因的KEGG富集分析显示,它们主要富集于苯丙氨酸代谢(图 4m)。这些结果有力地证明了NtMYB28是羟基肉桂酸生物合成的调控因子,并验证了GMRN的准确性。基于LF和HM条件下显著的温度差异,并考虑到MYB家族成员已知的调控作用,我们进行了低温胁迫实验,以研究NtMYB28在非生物胁迫响应中的功能。在冷胁迫下,NtMYB28-OE株系的生长优于野生型。与野生型植株相比,NtMYB28-OE株系的叶绿素含量更高,抗氧化能力也更强。相反,NtMYB28-KO株系在冷胁迫下表现出严重的生长抑制,且抗氧化活性显著降低。这些结果表明,NtMYB28也可作为烟草分子育种的候选抗逆基因。
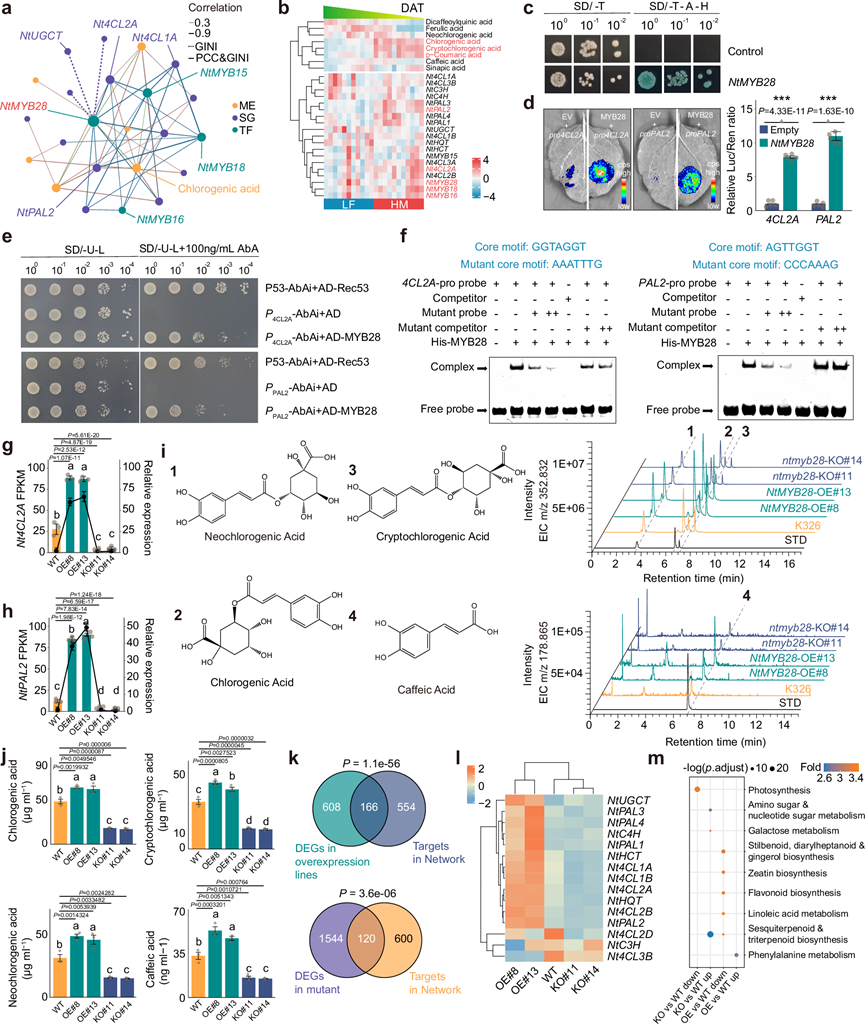
图4. 转录因子NtMYB28调节羟基肉桂酸的生物合成和参与苯丙素途径的基因的表达。
(a) 从绿原酸生物合成的GMRN中提取的子网络。(b) ME 、SG和TF的表达模式。(c) NtMYB28在酵母中的转录激活分析。(d) 瞬时表达分析显示NtMYB28对LUC报告基因(由Nt4CL2A和NtPAL2启动子驱动)的转录激活作用。(e) 使用诱饵和猎物或阴性对照,通过单杂交系统检测NtMYB28蛋白与Nt4CL2A和NtPAL2启动子的特异性结合。(f) 使用源自Nt4CL2A和NtPAL2启动子的探针进行NtMYB28蛋白的EMSA分析。(g-h) RNA-seq 和 RT-qPCR 结果表明,Nt4CL2A和NtPAL2受NtMYB28正向调控。(i) 比较野生型 (WT)、NtMYB28 过表达( OE) 和ntmyb28敲除 (KO) 烟草中绿原酸、新绿原酸、隐绿原酸和咖啡酸的质谱数据。(j) 烟草叶片中绿原酸、新绿原酸、隐绿原酸和咖啡酸的含量。(k) GMRN 能够准确预测NtMYB28的靶基因。(l) 苯丙素代谢途径相关基因的热图分析。(m) KEGG通路富集。
05
通过GMRN鉴定出一种未表征的转录因子,该因子调控脂质生物合成
在ERF-GMRN中,连接性最高的五个转录因子中,只有NtERF167的功能未知。NtERF167与许多参与脂质代谢的SG聚类。与此一致,GMRN的KEGG富集分析也显示,在VIII簇中多种脂质生物合成通路显著富集。因此,他们推测NtERF167可能是调控脂质代谢的关键转录因子。他们进一步从ERF-GMRN中提取了一个以这些脂质及其对应应激基因为中心的亚调控网络。NtERF167与脂质和应激基因表现出直接且强的相关性(图 5a)。脂质生物合成基因、相关转录因子的表达模式以及脂质的积累模式表现出高度一致性(图 5b)。系统发育分析表明,NtERF167与NtERF1和NtSHINE2(NtSHN2)相关。NtERF167的表达模式与长链酰基辅酶A合成酶2(NtLACS2)的表达模式高度相关且相似,并且在调控网络中,NtERF167和NtLACS2的重要性排名高于其他SG,表明NtERF167可能是NtLACS2的潜在调控因子(图 5b)。在GMRN中,与NtERF167共调控的基因和代谢物主要富集于脂质代谢和类黄酮生物合成途径。此外,NtERF167的 GMRN 中共表达基因的顺式元件主要包括 DREB 和 ERF TF 的结合位点。
转录激活活性分析和亚细胞定位表明,NtERF167具有TF的功能(图 5c-d)。NtLACS2 的启动子区域(ATG 上游 2000 bp 区域)包含ERF 结合基序 GCC-box。双荧光素酶报告基因检测、酵母单杂交 (Y1H) 检测和电泳迁移率变动分析 (EMSA) 表明,NtERF167 蛋白可直接结合NtLACS2的启动子(图 5e-g)。
为了验证NtERF167在脂质生物合成中的功能,他们构建了由35S启动子驱动的NtERF167过表达转基因烟草植株。此外,他们利用CRISPR/Cas9介导的基因组编辑技术,构建了与NtERF167过表达植株具有相同遗传背景的NtERF167功能缺失突变体。我们获得了两个独立的NtERF167过表达株系(NtERF167-OE#1和6)和两个独立的NtERF167突变株系(NtERF167-KO#3和28)。与野生型株系相比,过表达株系中NtLACS2的mRNA水平显著升高,而在NtERF167 -KO突变体中则显著下调(图 5h)。与WT植株相比,两个NtERF167过表达-OE株系的总脂质含量显著增加,而NtERF167-KO株系的总脂质含量则显著降低(图 5i)。PCA显示,两个OE株系和两个KO株系的脂质组学特征与WT植株明显不同。OE株系中大多数甘油磷脂(磷脂酸、磷脂酰甘油、磷脂酰肌醇、磷脂酰丝氨酸、溶血磷脂酰甘油和溶血磷脂酰乙醇胺)的含量显著高于WT或KO株系。此外,过表达植株中骨骼脂质DAG和TAG的含量更高(图 5j)。为了阐明NtERF167如何调控脂质生物合成,他们对NtERF167-OE、NtERF167-KO和WT植株进行了RNA-seq。WT、OE和KO株系间DEG与GMRN中预测的NtERF167靶基因的重叠度显著高于随机对照组(图 5k)。一系列参与游离脂肪酸(FFA)氧化和脂质代谢的重要基因在WT、OE和KO株系间存在差异表达(图 5l)。此外,对KO和OE株系间表达模式相反的DEG进行KEGG富集分析发现,它们主要富集于脂质代谢通路(图 5m),表明NtERF167可能与多个生物学通路相关。这些结果表明NtERF167是脂质代谢的关键调控因子,并且NtERF167的缺失会扰乱脂质代谢通量。他们还评估了NtERF167的耐寒性功能。与野生型相比,NtERF167功能缺失导致烟草对冷胁迫的敏感性增加。在低温条件下,NtERF167-KO株系的叶绿素含量显著降低,膜脂过氧化水平升高。这些结果表明NtERF167在非生物胁迫响应中也发挥作用。
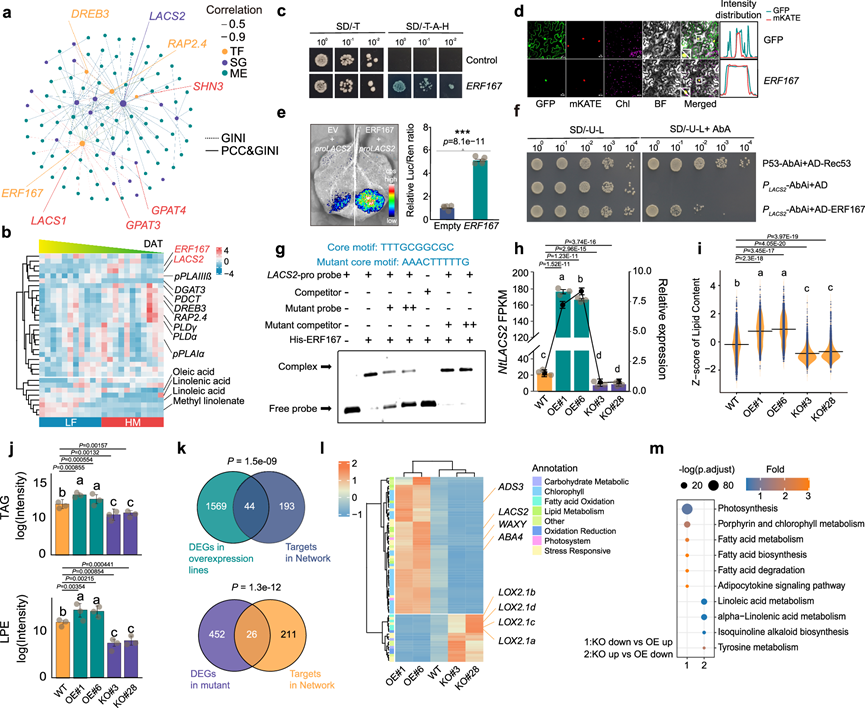
图5. NtERF167是脂质代谢的关键调控因子,会扰乱脂质组通量。
(a) 从GMRN中提取的脂质生物合成子网络。(b) 脂质、SG和TF的表达模式。(c) NtERF167在酵母中的转录激活分析。(d) NtERF167-GFP融合蛋白在烟草叶片细胞中的亚细胞定位。(e) 瞬时表达分析显示NtERF167对LUC报告基因(由NtLACS2启动子驱动)的转录激活。(f) 利用单杂交系统,以诱饵和猎物或阴性对照检测NtERF167与NtLACS2启动子的特异性结合。(g) 使用源自NtLACS2启动子的探针进行EMSA实验,检测NtERF167蛋白。(h) RNA-seq和RT-qPCR结果表明,NtLACS2受NtERF167正向调控。(i) 烟草叶片中的脂质含量。(j) 烟草叶片中的甘油三酯(TAG)和溶血磷脂酰乙醇胺(LPE)含量。(k) GMRN能够准确预测NtERF167的靶标。(l) 基因表达模式在NtERF167过表达 (OE) 和NtERF167敲除 (KO) 烟草中呈现相反的表达模式。(m) KEGG基因富集分析显示NtERF167过表达 (OE) 和NtERF167敲除 (KO) 烟草中呈现相反的表达模式。
06
GMRN图展示了香气形成的关键调控因子
植物香气是由多种芳香挥发性代谢物组成的复杂组合,主要来源于植物体内类胡萝卜素的降解。脂氧合酶(LOX)催化多不饱和脂肪酸(如亚油酸和亚麻酸)的氧化。脂肪酸氧化中间步骤产生的自由基会导致类胡萝卜素色素的氧化降解。LOX催化的共氧化反应涉及类胡萝卜素的非特异性裂解,产生脱辅基类胡萝卜素挥发物。为了鉴定调控类胡萝卜素降解和香气形成的基因,他们重点研究了DHA,它是植物中一种重要的风味物质和信号分子。GMRN分析表明,NtCYC可能通过调控NtLOX2的表达来调控DHA的生成。系统发育分析揭示了拟南芥和烟草中NtCYC与TCP4之间存在密切的进化关系(补充图 31)。以几种香气萜类代谢物为中心的子网络揭示了参与萜类代谢的多种代谢物种类和转录因子家族,表明萜类生物合成过程的复杂性(图 6a)。在该网络中,DHA、LOX和大多数TCP均位于簇VI中。对GMRN进行KEGG富集分析表明,可能与香气形成相关的代谢通路,例如FFA降解、谷胱甘肽代谢、氧化磷酸化、倍半萜和三萜生物合成,在聚类VI中特异性富集(图 2c)。由于LOX基因也参与茉莉酸(JA)的合成,我们检测了参与类胡萝卜素降解或JA合成的基因及其相关转录因子的表达模式,以及DHA的积累模式。NtCYC的表达模式与NtLOX2和DHA的表达模式高度相关且相似(图 6b),表明NtCYC可能是NtLOX2的潜在调控因子。
转录激活活性分析和亚细胞定位结果表明,NtCYC具有TF的功能(图 6c-d)。Y1H、电泳迁移率变动分析(EMSA)和双荧光素酶报告基因检测表明,NtCYC 蛋白能够结合NtLOX2启动子(图 6e-g)。为了进一步研究NtCYC在香气形成和类胡萝卜素代谢中的功能,他们构建了由35S启动子驱动的NtCYC过表达转基因烟草植株。此外,利用 CRISPR/Cas9 介导的基因组编辑技术构建了NtCYC功能缺失突变体。我们获得了两个独立的NtCYC过表达株系(NtCYC-OE#7 和 12)和两个独立的NtCYC突变株系(NtCYC-KO#27 和 43)。他们对烟草叶片提取物进行了香气代谢物分析,包括β-紫罗兰酮、β-大马酮和DHA。由于烟草中香气化合物的浓度相对较低,使用GC-MS检测存在挑战,因此我们选择灵敏度更高的LC-MS进行分析。在两个NtCYC过表达株系的叶片中,DHA和β-紫罗兰酮的含量显著高于野生型植株,而在NtCYC敲除突变体的叶片中,其含量则显著低于野生型植株(图 6h-j )。NtLOX2的mRNA水平在过表达株系中显著高于野生型,而在NtCYC敲除突变体中则下调(图 6k)。此外,由于脂氧合酶(LOX)通过游离脂肪酸(FFA)氧化降解类胡萝卜素生成香气化合物,他们检测了NtCYC过表达(NtCYC-OE)、NtCYC敲除(NtCYC-KO)和WT植物中丙二醛(MDA)的含量——MDA是脂质氧化的重要指标。结果表明,与WT植物相比,NtCYC -OE植物中MDA含量显著增加,而ntcyc -KO植物中MDA含量则显著降低(图 6l)。有趣的是,β-大马酮的含量与DHA和β-紫罗兰酮的含量呈现相反的结果(图 6i)。由于β-大马酮不受LOX降解,NtCYC介导的NtLOX2表达增强将代谢流转向由LOX催化的DHA和β-紫罗兰酮。
为了阐明NtCYC如何调控香气形成,他们对NtCYC-OE、NtCYC-KO和WT植物进行了RNA-seq。PCA结果显示,两个OE株系或两个KO株系分别具有相似的转录组,且与WT株系明显不同。WT、OE和KO植物中DEG与GMRN中预测的NtCYC靶基因的重叠度显著高于随机对照组(图 6m)。此外,用于构建茉莉酸(JA)生物合成亚调控网络的SG在WT、OE和KO株系中的表达水平也存在差异(图 6n)。对这些DEG进行GO和KEGG富集分析,揭示了与GMRN的KEGG富集分析相似的分子功能,例如参与倍半萜和三萜的生物合成。总之,NtCYC是香气形成的一个调控因子,并影响萜类代谢的代谢通量。鉴于NtCYC可能介导氧化稳态,并且有证据表明某些CYC转录因子在非生物胁迫中发挥负调控作用,他们研究了NtCYC在冷胁迫下的作用。与WT相比,NtCYC-KO)植株在冷胁迫下表现出更强的生长表型。NtCYC-KO株系的叶绿素含量更高,同时过氧化氢积累和膜脂过氧化水平显著降低。这些结果表明,NtCYC通过过氧化物介导的机制负向调节烟草的耐寒性。
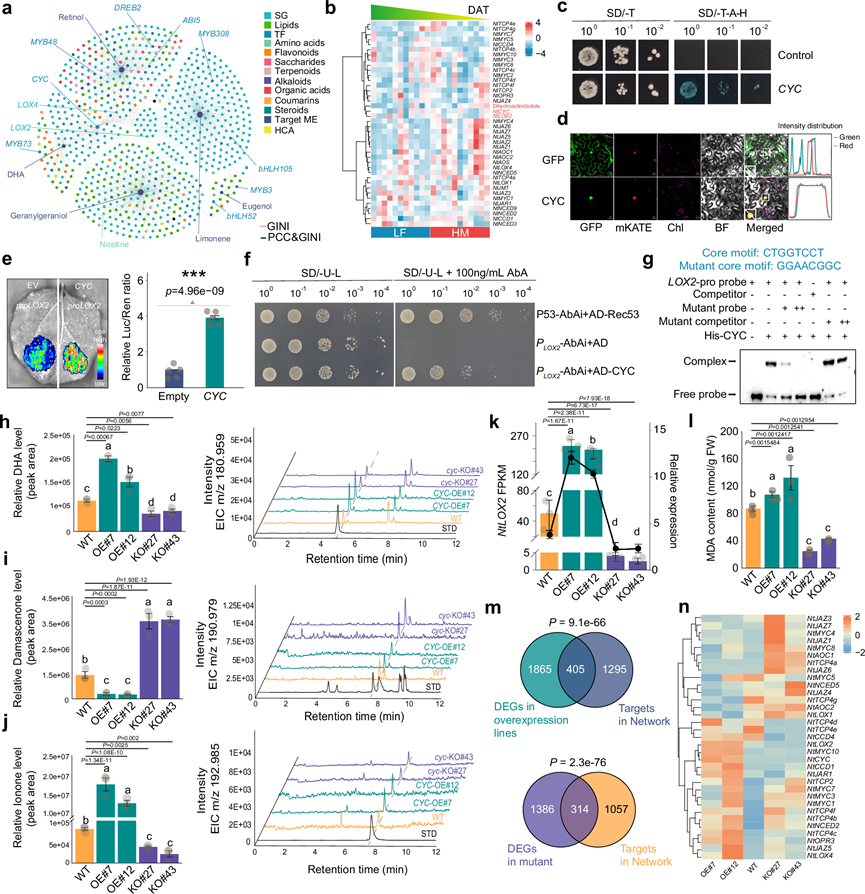
图6. 转录因子 NtCYC 改变了香气的形成过程。
(a) 从GMRN中提取的类胡萝卜素降解子网络。(b) 代谢物、SG和TF的表达模式。“4”和“-4”分别代表Z评分标准化表达的最大值和最小值。(c) NtCYC在酵母中的转录激活分析。(d) NtCYC -GFP融合蛋白在烟草叶片细胞中的亚细胞定位。(e) 瞬时表达分析显示NtCYC对LUC报告基因(由NtLOX2启动子驱动)的转录激活。(f) 利用单杂交系统,以诱饵和猎物或阴性对照(诱饵/pGADT7)检测NtCYC蛋白与NtLOX2启动子的特异性结合。(g) 使用源自NtLOX2启动子的探针进行EMSA实验,检测NtCYC蛋白。(h-j) 烟草叶片中二氢猕猴桃内酯、大马酮和紫罗兰酮的含量及其相应的质谱数据。(k) RNA-seq和RT-qPCR结果表明NtLOX2受NtCYC正调控。(l) 烟草中丙二醛(MDA)含量的比较。(m) GMRN能够准确预测NtCYC的靶标。(n) 野生型 (WT)、 NtCYC 过表达 (NtCYC -OE) 和NtCYC敲除(NtCYC -KO) 烟草中类胡萝卜素通路相关基因的表达模式。
+ + + + + + + + + + +
结 论
本研究通过整合来自两个生态环境迥异的地区田间种植烟草叶片的动态转录组和代谢组数据,构建了一个基因组规模的代谢调控网络。利用多算法整合方法,将25984个基因和633个代谢物映射到317万个调控对中。该网络揭示了三个关键的转录枢纽,包括NtMYB28 (通过调控Nt4CL2和NtPAL2的表达促进羟基肉桂酸的合成)、NtERF167 (通过激活NtLACS2增强脂质合成)和NtCYC (通过诱导NtLOX2驱动香气物质的产生)。这些转录枢纽通过重塑代谢流,显著提高了目标代谢物的产量。本研究提供了烟草代谢调控的系统级图谱,并可能有助于指导代谢工程。
+ + + + +



