

 English
English文献解读|Nat Commun(15.7):对乳腺癌异质性的综合空间蛋白质组学分析揭示了癌细胞的表型可塑性
✦ +
+
论文ID
原名:Integrated spatial proteomic analysis of breast cancer heterogeneity unravels cancer cell phenotypic plasticity
译名:对乳腺癌异质性的综合空间蛋白质组学分析揭示了癌细胞的表型可塑性
期刊:Nature Communications
影响因子:15.7
发表时间:2025.11.25
DOI号:10.1038/s41467-025-65477-6
背 景
乳腺癌原发肿瘤异质性是导致耐药和复发的驱动因素,并影响免疫逃逸和肿瘤进展。尽管肿瘤内异质性已在基因组水平上得到广泛研究,但其功能后果以及与肿瘤微环境的相互作用仍未得到充分探索。相反,异质性的功能后果及其与肿瘤微环境的相互作用尚未得到充分研究。
实验设计
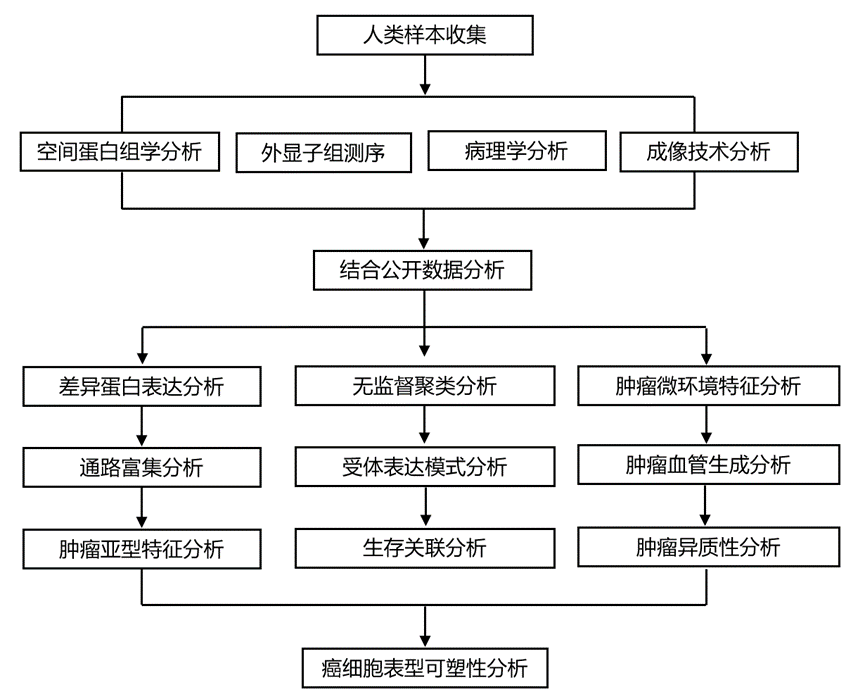
结 果
01

空间蛋白质组学重现了肿瘤亚型和分级之间的已知差异
为了全面分析乳腺癌中蛋白质组的异质性,研究团队开发了一个分析框架,用于检测单个肿瘤内具有不同组织病理学特征的区域的蛋白质组组成(图 1A)。他们分析了来自33例乳腺肿瘤队列的280个显微切割的肿瘤区域。每个肿瘤组织块的切片均进行了三种关键乳腺癌受体的染色:雌激素受体(ER)、孕激素受体(PR)、HER2(ERBB2)和泛细胞角蛋白(panCK)。基于受体表达谱,他们利用人工智能进行细胞定量和病理图谱分析,确定了肿瘤内的不同区域(图1A)。随后,对每个区域进行激光捕获显微切割,并作为独立的样本进行分析。这些样本包括19个正常邻近组织样本、12个导管原位癌(DCIS)样本、10个1级肿瘤区域、139个2级肿瘤区域和100个3级肿瘤区域。
作为对数据的初步评估,他们进行了一系列统计检验,以比较肿瘤分级和亚型。超过2400种蛋白质在癌组织和邻近正常组织之间存在显著差异。正如预期的那样,癌组织中增殖标志物(如KI67、PCNA和DNA聚合酶)以及代谢酶(如SCD、PYCR2、GLUL)的水平升高。富集分析显示,癌组织样本中核苷酸生物合成通路、DNA修复、染色体组织和线粒体翻译的表达水平较高,而正常对照组中脂蛋白代谢、细胞黏附和体液免疫反应的表达水平较高。与已知的3级肿瘤增殖率较高相一致,比较3级和2级肿瘤的蛋白质组发现,3级肿瘤中HR+和TNBC区域的PCNA、MCM蛋白和核苷酸代谢相关蛋白均高表达(图 1B),而细胞周期调节因子WEE1的表达水平较低。亚型分离的富集分析显示,3级TNBC也高表达MCM复合物蛋白、DNA复制相关蛋白、嘧啶代谢相关蛋白以及抗原加工和呈递相关蛋白(图 1C)。HR+ 3级肿瘤也高表达抗原加工和呈递相关蛋白、蛋白酶体复合物蛋白以及参与DNA生物合成的蛋白(图 1D)。
亚型比较显示,TNBC 区域一碳代谢通路蛋白(PHGDH 和 PSAT)表达较高,同时 PODXL 水平也升高。PODXL 是一种细胞表面抗黏附性唾液酸黏蛋白,可促进肿瘤细胞脱落并增强其迁移能力(图 1E)。HR+ 区域 AGR2 和 AGR3(均为已知的雌激素受体 (ER) 相关蛋白)以及 KRT18 和 KRT19(HR+ 乳腺癌的典型标志物)的水平也升高。分级分离的富集分析显示,3 级肿瘤的差异更为显著,TNBC 区域核糖体和染色质蛋白富集,而 HR+ 区域 PI3K 和 I 型干扰素水平较高(图 1F-G)。总而言之,这些初步比较为基于区域的空间方法提供了概念验证,因为重现了癌症发展和进展以及癌症亚型之间的已知变化,并提供了可进一步研究的蛋白质组范围变化的资源。
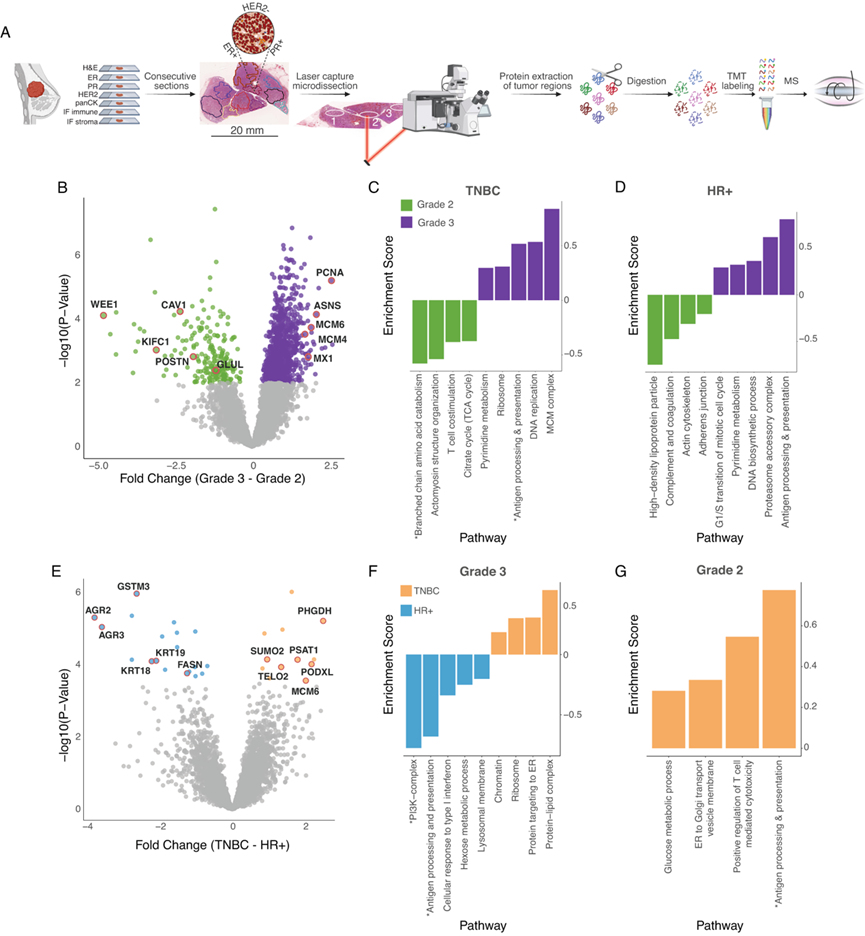
图1. 基于多区域 MS 的蛋白质组学阐明了与肿瘤组织病理学相关的蛋白质组学差异。
(A) 多区域蛋白质组学工作流程示意图。(B) 火山图显示了HR+和TNBC区域中2级和3级之间显著变化的蛋白质。(C-D) 在三阴性乳腺癌(TNBC)肿瘤区域和激素受体阳性(HR+)肿瘤区域中鉴定的富集通路。(E) 火山图显示了TNBC和HR+之间显著变化的蛋白质。(F) 条形图显示了3级HR+和TNBC的富集分析。(G) 条形图显示了2级HR+和TNBC的富集分析。
02
在高级别肿瘤中,受体异质性降低,而蛋白质组异质性升高
为了揭示癌症异质性的驱动因素,他们基于肿瘤区域的整体蛋白质组谱进行了无监督聚类分析。分析结果显示,肿瘤分级、受体表达模式和患者来源构成了一个明显的层级分离。肿瘤分级是最显著的区分因素,主要区分了2级和3级肿瘤(图 2A)。在二级层级上,受体表达模式有效地将HR+、TNBC和HER2+区域进行了分层(图 2B)。三级层级的分层与患者来源相关,来自同一患者的区域通常聚集在一起(图 2C)。然而,患者间的差异可能部分受到TMT组和实验批次的影响。因此,他们将分析重点放在肿瘤内部的比较上,即比较同一TMT组和同一实验批次内的样本。有趣的是,他们发现多个TNBC区域与HR+肿瘤聚集在一起,表明单个肿瘤内受体表达存在异质性。虽然一些肿瘤,特别是2级肿瘤,表现出受体多样性,但3级肿瘤则表现出更高的受体同质性(图 2A-C)。
为了确定与肿瘤分级相关的受体异质性的统计学意义,他们定义了一个名为受体-ITH的肿瘤内受体异质性评分。该评分考虑了单个肿瘤中共存的不同受体表达模式的数量以及最主要模式的比例(图 2D)。因此,当共存的模式更多且分布更均匀时,受体-ITH值越高。2级和3级肿瘤的受体-ITH比较显示,随着肿瘤进展,肿瘤内异质性有降低的趋势(图 2E)。由于本研究队列中的肿瘤数量限制了统计效力,他们使用来自一个包含385例乳腺肿瘤的独立队列的成像质谱流式细胞术数据验证了本研究的结果。通过检测HR+和TNBC细胞,他们分析了香农多样性指数,该指数基于不同细胞群的数量及其相对分布来量化多样性。与他们的结果一致,对385例肿瘤的检查显示,高级别肿瘤的受体多样性显著低于低级别肿瘤(图 2F)。这些结果提示,即使在治疗前,原发部位的肿瘤也可能存在改变肿瘤亚型的潜在选择压力。
聚类结果表明受体表达模式与整体蛋白质组模式之间存在不一致,因为基于蛋白质的聚类通常包含具有不同受体表达的区域(图 2C)。为了量化这些差异,他们基于成对蛋白质的Pearson相关性定义了蛋白质组肿瘤内异质性评分,即P-ITH评分(图 2G)。令人惊讶的是,与受体-肿瘤内异质性(Receptor-ITH)随肿瘤分级升高而降低的情况相比,P-ITH在3级肿瘤中显著高于2级肿瘤(图 2H)。全局Pearson相关性分析显示,受体-肿瘤内异质性与无病生存期(DFS)呈显著正相关,而与肿瘤分级呈负相关。相反,P-ITH 与肿瘤分级和平均肿瘤细胞密度呈正相关。与这些观察结果一致,P-ITH 与无病生存期(DFS)呈负相关,并且在发生转移或复发的患者中升高(图 2I)。总之,这些结果强调了肿瘤功能(由整体蛋白质组谱反映)与受体表达模式存在差异,为深入了解这些因素在肿瘤发生过程中的复杂相互作用提供了宝贵的见解。
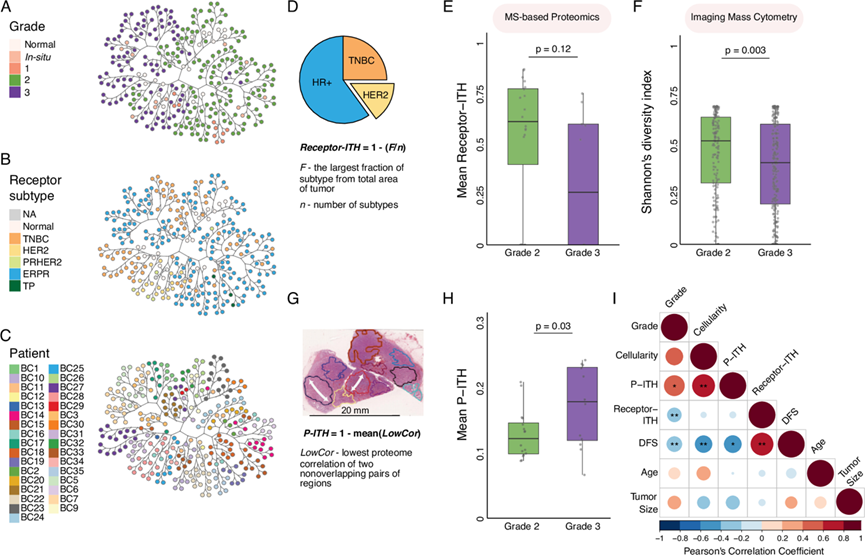
图2. 在高级别肿瘤中,受体-ITH 降低,而蛋白质组-ITH 升高。
(A) 基于改进的最小方差方法,采用树叶模型对肿瘤区域进行无监督聚类,其中每个节点代表一个肿瘤区域。图中所示的树状图根据肿瘤分级进行颜色编码。(B) 图 (A)相同,但颜色基于受体表达。(C) 与图(A)相同,但颜色基于患者身份。(D) 基于免疫组织化学(IHC)染色并按肿瘤计算的受体-肿瘤内异质性评分(Receptor-ITH)示意图。(E) 肿瘤中 Receptor-ITH 评分的分布。(F) 箱形图显示了METABRIC队列中2级(n = 141)和3级(n = 221)肿瘤的Shannon多样性指数分布。(G) 计算每个肿瘤/ TMT集的蛋白质组学肿瘤内异质性评分(P-ITH)的示意图。(H) 箱线图,显示2级和3级肿瘤中 P-ITH 评分的分布。(I) 肿瘤特征相关性热图。
03
P-ITH受肿瘤微环境相互作用的影响
为了揭示P-ITH的根本原因,他们首先研究了基因组ITH对下游蛋白质组学特征的潜在影响。他们使用连续的FFPE肿瘤切片,对与蛋白质组学分析相同的区域进行激光显微切割,并进行外显子组测序。整合外显子组数据库,他们对从4个HR+肿瘤中提取的47个肿瘤区域中的5227个基因进行了分析。变异检测结果与其他乳腺癌基因组数据集的结果相当。与之前的研究一致,患者间的差异比同一患者不同区域间的差异更为显著。绝大多数突变基因具有患者特异性。然而,在单个患者的所有区域均存在或在患者间普遍存在的基因包括COSMIC数据库中收录的已知乳腺癌驱动基因突变,例如PIK3CA和TP53。为了进一步检验蛋白质组异质性是否与基因组异质性相对应,他们绘制了每个肿瘤内各区域间两两排序的余弦相似度(图 3A-D)。
鉴于基因组异质性并未直接影响蛋白质组异质性(P-ITH),他们利用蛋白质组数据,探索了P-ITH的其他驱动因素,以识别能够区分同一患者多个肿瘤不同区域的蛋白质。对于每种蛋白质,他们计算了每位患者的中位绝对偏差(MAD)值,从而确定变异性最高和最低的蛋白质,分别对应于最高和最低的MAD值。对最保守蛋白质的富集分析显示,其参与了与翻译、蛋白质折叠、周转和转运相关的过程(图 3E)。他们将这些蛋白质定义为不受空间可塑性影响的核心癌症蛋白质组;另一方面,对变异性最高的蛋白质的富集分析则鉴定出多种免疫相关过程,主要涉及抗原呈递(例如HLA分子)、组蛋白、可能反映血管邻近性的蛋白质(例如HBB)以及代谢过程(图 3F)。这些结果表明,恒定的癌症蛋白质组主要由反映癌细胞基本增殖和生物合成需求的内在因子构成。相比之下,异质性蛋白质组则反映了由与肿瘤微环境(TME)相互作用塑造的细胞可塑性。
鉴定出高度可变的蛋白质凸显了肿瘤微环境(TME)相互作用对蛋白质组的潜在影响。为了研究肿瘤区域蛋白质组与TME特征之间的联系,他们对TME成分进行了多重成像,包括T细胞(CD3+)、巨噬细胞(CD68+)、成纤维细胞(αSMA+)、内皮细胞(CD31+)和癌细胞(panCK)(图 4A)。他们开发了一个计算流程,将H&E染色和免疫组化(IHC)染色组织切片图像中的蛋白质组感兴趣区域(P-ROI)转移到连续的免疫荧光图像上。然后,他们开发了两个基于深度学习的分析流程,用于量化细胞成分和血管。这些分析在三个层面进行:整个肿瘤切片、P-ROI以及P-ROI周围的邻域(图 4B)。
他们总共对来自队列中11例患者的47个肿瘤切片进行了空间分析,涵盖了121个蛋白质组区域及其邻近区域。首先,他们定量了细胞数量和血管数量,而不考虑蛋白质组感兴趣区域(P-ROI),以评估所有肿瘤微环境(TME)成分的参与情况。在切片水平上对T细胞、巨噬细胞、血管和成纤维细胞进行定量分析,揭示了T细胞浸润水平的广泛差异,从无T细胞的“冷”肿瘤到CD3阳性细胞比例>40%的肿瘤(图 4C)。整个切片中巨噬细胞(CD68+)的数量变化范围为0.5%至18%,而成纤维细胞(αSMA+)的数量变化范围最大,从2.4%到64.7%(图4D-E)。他们计算的血管面积显示,平均占总组织面积的1-2%,仅有两个组织切片显示出更高的百分比(图 4F)。对这些肿瘤微环境(TME)成分之间相关性的分析揭示了一个总体趋势:免疫成分,例如CD68+巨噬细胞和CD3+ T细胞,呈正相关,尽管这种相关性不具有统计学意义。然而,泛细胞角蛋白阳性(panCK+)上皮细胞与巨噬细胞和T细胞均呈显著负相关。相反,αSMA+成纤维细胞与CD3+ T细胞呈负相关(图 4G)。此外,血管(BV)与成纤维细胞之间也观察到正相关趋势。这些分析再次证实了先前发表的癌症相关成纤维细胞(CAFs)对血管生长的积极影响以及组织纤维化与免疫浸润之间的负相关性。他们发现免疫浸润与血管之间无显著相关性,这可能是由于血管百分比值的分布存在偏斜所致。然而,当将肿瘤分为血管丰富(BV-high)组和血管稀少(BV-low)组时,与BV-high区域相比,BV-low区域的T细胞和巨噬细胞数量显著更高(图 4H)。这一观察结果提示,T细胞和巨噬细胞可能募集到血管较少、缺氧程度更高的区域。有趣的是,按癌症分级分析免疫浸润情况发现,3级肿瘤中T细胞和巨噬细胞水平高于2级肿瘤,同时成纤维细胞数量较少(图 4I)。他们利用358例患者的成像质谱流式细胞分析数据进一步验证了这些结果,结果显示,肿瘤分级越高,免疫浸润程度越深,成纤维细胞越弱(图 4J)。这些有趣的发现表明,癌症相关成纤维细胞数量随癌症进展而减少,而免疫浸润则随肿瘤进展和多样化而增加。
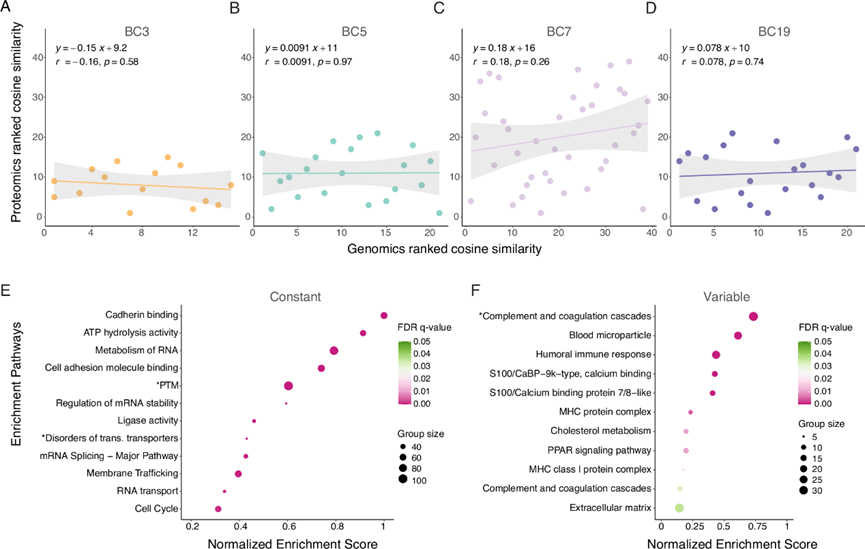
图3. ITH 的蛋白质基因组分析。
(A-D) 展示了四位ER阳性乳腺癌患者各肿瘤区域内基因组和蛋白质组间排序余弦相似性的成对相关性。(E-F) 通路富集分析。
04
TME分析将蛋白质组图数据与肿瘤血管生成联系起来
为了更好地理解癌症与TME的相互作用,他们深入研究了区域层面的这些关联,建立了肿瘤空间表型与蛋白质组图谱之间的联系。由于P-ROI区域内血管稀少,因此他们对血管进行了区域定量分析,包括P-ROI区域及其周围500 µm的环状区域。为了鉴定与血管密度(BV%)相关的蛋白质,他们进行了Pearson相关性分析,并鉴定出1103个与BV%显著相关的蛋白质,其中95%呈负相关。在这些蛋白质中,他们发现了PIK3CA、IL13RA1、LIMK1、SLC16A1、SOX4、YBX2、MKI67、SCD、CDK1和ENO2,这些蛋白质已知与缺氧或乳腺癌相关(图 5A)。 PIK3CA是乳腺癌中常见的突变或扩增基因,其与BV%呈显著正相关。PIK3CA与癌细胞存活、转移和血管生成密切相关。本研究中PIK3CA与BV%的相关性提示其可能参与肿瘤血管生成。同样,IL13RA1也与BV%呈显著正相关(图 5A)。与此一致的是,既往研究表明IL13RA1在缺氧条件下表达下调。LIMK1已证实参与人乳腺癌细胞的肿瘤血管生成,其在富含BV的区域表达上调。相反,绝大多数此类蛋白与BV%呈负相关,提示缺氧或TME中血管减少可能是其表达上调的一种适应性反应,以应对氧气和营养匮乏。在这些蛋白质中,他们发现了增殖细胞的标志性蛋白MKI67和有丝分裂细胞周期调节因子CDK1。SOX4是一种转录因子,已知其是上皮-间质转化(EMT)的调节因子,已证实能够促进乳腺癌的生长和转移。从代谢角度来看,他们观察到在低血管密度(BV-low)区域,糖酵解酶ENO2和乳酸转运蛋白SLC16A1(两者均为瓦博格效应的关键组成部分)的表达上调,这与预期的低氧代谢适应相一致。与这些变化相符的是,他们检测到SCD表达增加,SCD是脂质生物合成的关键酶,能够支持快速增殖细胞的膜形成和能量储存。因此,肿瘤内的血管化和低氧微环境具有不同的蛋白质组学特征,这些特征与肿瘤进展和治疗反应密切相关。
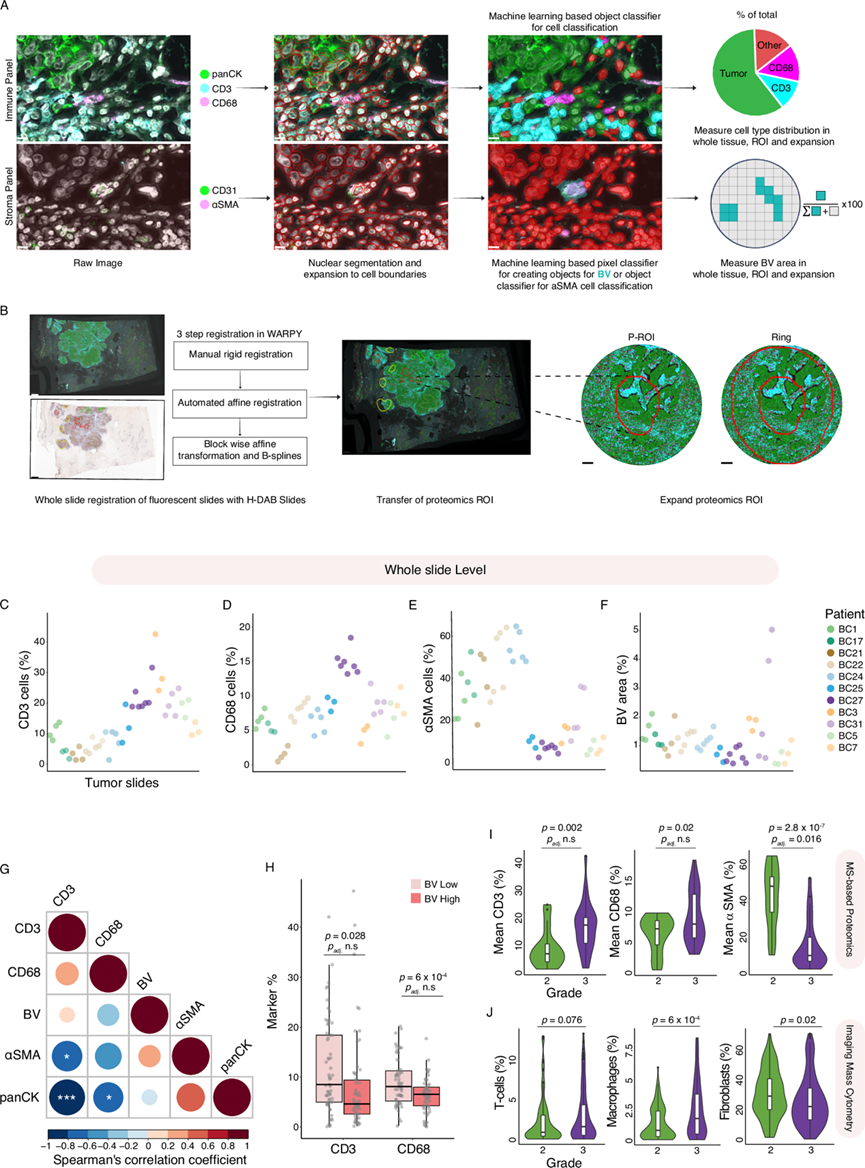
图4. 基于多重成像的 TME 成分全切片分析。
(A) 多重成像工作流程示意图。(B) 多层蛋白质组学和成像整合的分析流程示意图。(C-F) 散点图显示了全切片图像中肿瘤微环境 (TME) 成分的定量结果。(G) 全组织分析中 CD3+ 细胞、CD68+ 细胞、BV、αSMA+ 细胞和泛细胞角蛋白 (panCK+) 癌细胞百分比的相关性热图。(H) 箱线图描绘了 BV 低表达区和 BV 高表达区中 CD3+ T 细胞和 CD68+ 巨噬细胞百分比的分布。(I) 小提琴图结合箱形图显示了CD3+、CD68+和αSMA+细胞百分比在2级和3级肿瘤中的分布。(J) 小提琴图结合盒图显示了影像学质谱细胞数据集中每例METABRIC患者的平均T细胞、成纤维细胞和巨噬细胞百分比的分布。
05
四种癌症免疫表型的蛋白质组学分析
既往在乳腺癌和其他癌症类型中的研究描述了三种与T细胞相关的免疫表型:T细胞浸润型(热肿瘤)、冷肿瘤(缺乏T细胞)以及T细胞排除型或分隔型肿瘤,其中大量T细胞位于大型癌细胞聚类之外。在区域水平上分析免疫表型可以将其与每种表型的蛋白质组图谱联系起来。为此,他们对每个蛋白质组区域内的T细胞和巨噬细胞进行了定量,并额外测量了围绕蛋白质组感兴趣区域(P-ROI)的环状区域内的细胞丰度,以区分冷肿瘤和免疫细胞排除区域。他们对121个区域及其周围环状区域应用了相同的分析流程,分别针对T细胞和巨噬细胞进行分析。结合T细胞和巨噬细胞的表型,他们得到了四种主要的免疫表型:T细胞和巨噬细胞浸润(“浸润”)、T细胞和巨噬细胞未浸润(“冷”)、T细胞分隔且巨噬细胞浸润以及T细胞未浸润且巨噬细胞浸润(图 5B-C)。他们发现几乎所有免疫浸润区域都包含巨噬细胞,但并非所有区域都包含T细胞。为了鉴定免疫表型的蛋白质组学决定因素,他们首先进行了三因素方差分析(ANOVA),以检测显著变化的蛋白质,同时校正了肿瘤分级和受体表达的混杂因素(图 5D)。他们发现414种蛋白质与免疫亚型显著相关,但不受肿瘤分级和亚型的混杂影响。为了检验T细胞和巨噬细胞来源蛋白质的潜在贡献,他们将与免疫表型显著相关的2289种蛋白质(不考虑肿瘤分级和亚型)与这些细胞类型的公开特征蛋白进行了比较。在T细胞特征蛋白中,只有不到0.8%(19种蛋白质)出现在免疫表型显著列表中。同样,在巨噬细胞特征蛋白中,也只有不到6.8%(155种蛋白质)包含在免疫表型显著列表中。有趣的是,与冷区相比,免疫浸润区域中只有 94 种巨噬细胞特征蛋白升高,而 T 细胞特征蛋白均未升高。这些结果表明,与免疫差异相关的蛋白质组学信号中,只有不到 7.6% 来自免疫细胞本身,而其余信号可能反映了免疫细胞对癌细胞的影响。
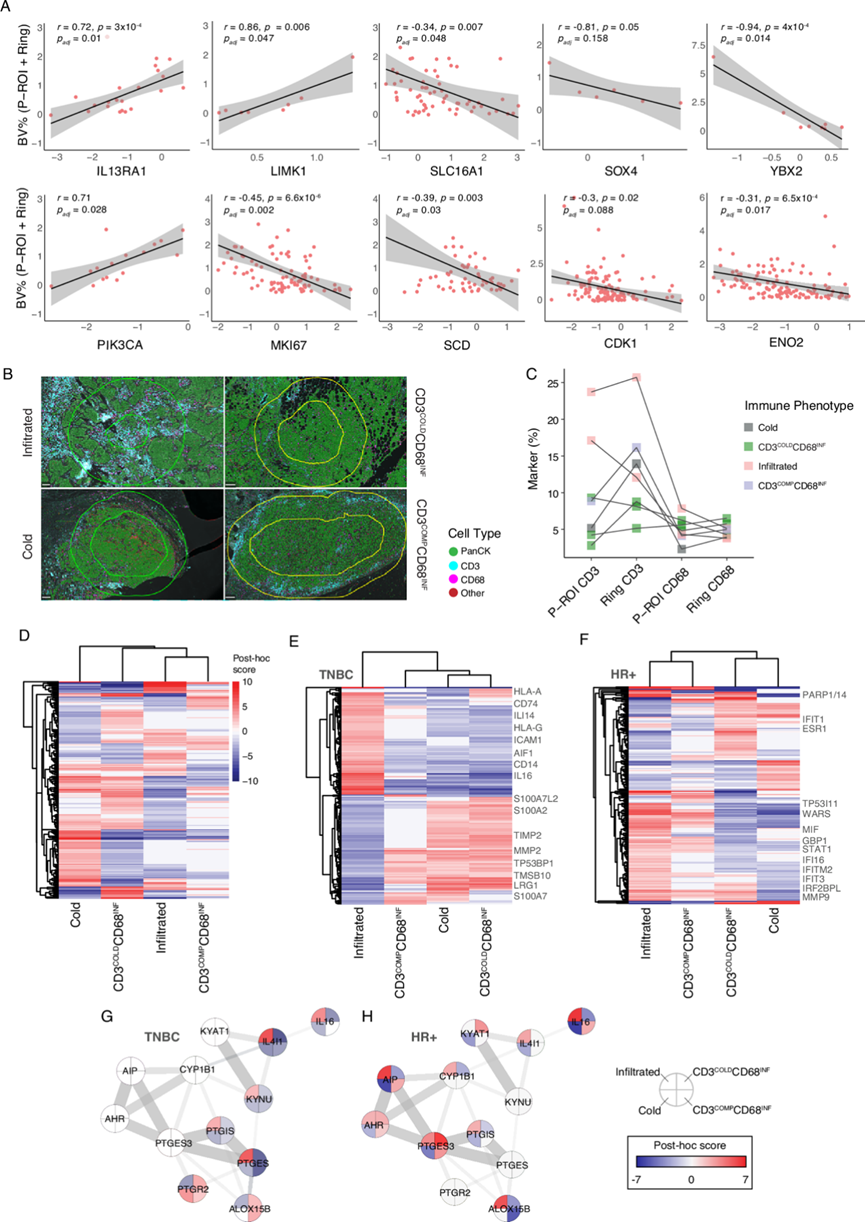
图5. 空间蛋白质组学与多重成像相结合,揭示了独特的 TME 相互作用。
(A) 散点图显示了P-ROI及其周围环形区域中蛋白表达值与血管百分比之间的Pearson相关性。(B) 免疫荧光分析。(C) 基于P-ROI及其周围环状区域内CD3+ T细胞和CD68+巨噬细胞百分比的BC1免疫表型散点图示例。(D) 414个与免疫表型相关的显著变化蛋白的热图。(E) TNBC P-ROI中658个免疫表型间显著变化蛋白的热图。(F) HR+ P-ROI中2822个免疫表型间显著变化蛋白的热图。(G-H) 犬尿氨酸通路、芳烃受体 (AHR) 和前列腺素的蛋白质网络图。
+ + + + + + + + + + +
结 论
本研究整合了280个肿瘤区域的多区域空间质谱蛋白质组学数据、外显子组测序数据和成像数据,以研究乳腺癌的空间蛋白质组异质性。随着肿瘤进展,蛋白质组异质性增加,这种增加独立于基因组异质性,但与微环境差异密切相关。结合免疫和基质成像,本研究揭示了一种动态的相互作用:低级别肿瘤表现出免疫浸润受限,而随着肿瘤级别升高,巨噬细胞和T细胞浸润水平显著提升。然而,在浸润区域,涉及犬尿氨酸和前列腺素的抗炎通路表达水平更高,表明其抗肿瘤活性受到抑制。与蛋白质网络的整合为乳腺癌免疫逃逸的潜在靶向介质提供了可能,这可为未来开发个性化乳腺癌疗法奠定基础。
+ + + + +



