

 English
English文献解读|Cancer Discov(33.3):前列腺癌异质性的生殖细胞和体细胞起源
✦ +
+
论文ID
原名:The Germline and Somatic Origins of Prostate Cancer Heterogeneity
译名:前列腺癌异质性的生殖细胞和体细胞起源
期刊:Cancer Discovery
影响因子:33.3
发表时间:2025.05.02
DOI号:10.1158/2159-8290.CD-23-0882.
背 景
前列腺癌是男性中最常见的内源性恶性肿瘤,随着人口预期寿命的延长,其发病率也在急剧上升。很大一部分前列腺肿瘤在临床上属于惰性肿瘤,无需根治性治疗,但也有一部分肿瘤具有侵袭性临床行为,可转移至远处,从而导致死亡。临床医生根据临床病理特征区分惰性肿瘤和侵袭性肿瘤,包括治疗前血清前列腺特异性抗原(PSA)浓度、肿瘤分级以及肿瘤大小和范围(T 类)。肿瘤分级是局部疾病致死率的最强预测指标,由泌尿生殖系统 (GU) 专家病理学家通过目视检查腺体结构和形态来确定。根据国际泌尿病理学会 (ISUP) 分级组 (GG) 系统,肿瘤分为五个等级,该系统是著名的 Gleason 分级系统的现代更新版本。 ISUP GG 1 肿瘤转移潜能极小,而 ISUP GG 5 肿瘤分化较差,播散风险显著增加,总体预后较差。前列腺肿瘤如何进化成不同的等级仍不清楚,越来越多的证据表明,种系和体细胞遗传学都会影响前列腺癌的演化。
实验设计
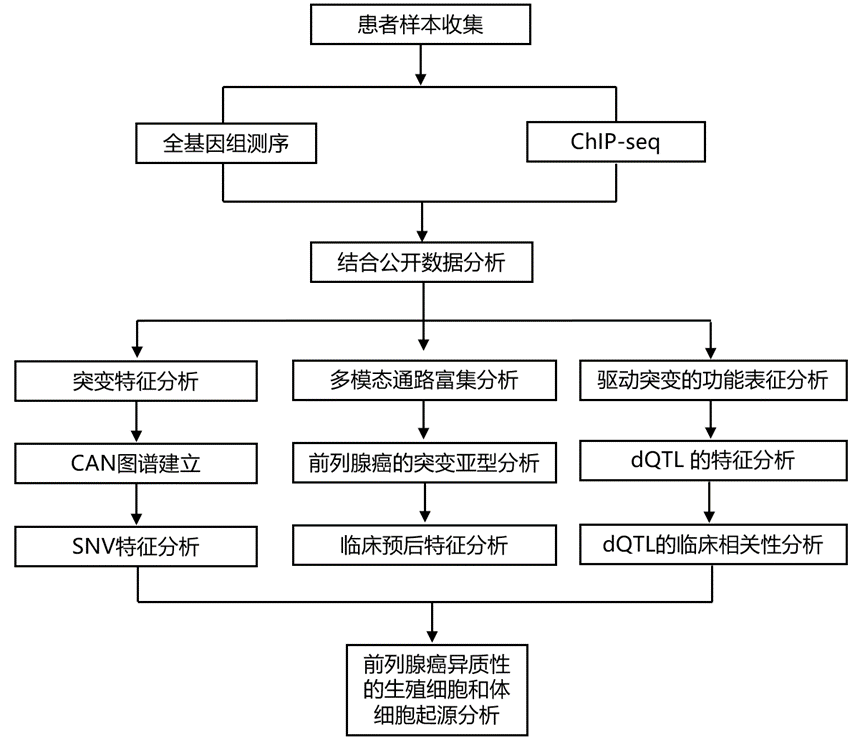
结 果
01

局限性前列腺癌的突变密度
为了阐明前列腺癌等级的突变和进化驱动因素,研究团队利用全基因组测序(WGS)技术研究了 666 例原发性局限性前列腺肿瘤(图 1A)。他们创建了 223 个驱动区域的图谱,其中大多数是由靶向测序无法检测到的结构变异引起的。许多驱动区域受到多种突变类型的改变;例如,FOXA1在 5.8% 的肿瘤中存在点突变,但在另外 10.1% 的肿瘤中以其他方式发生突变。利用三维染色质结构和增强子分析,他们鉴定出 35 个可预测特定前列腺癌驱动区域突变的种系 SNP。其中 11 个在 1991 名患者的Meta分析中得到了验证。十个驱动区域在高级别癌症中更频繁地发生突变,这些发生在肿瘤发展的早期并且与更差的临床结果相关(图 1A)。
正如预期的那样,前列腺肿瘤含有少量体细胞单核苷酸变异(SNV),核基因组测序的中位数为 0.42 SNV/Mbp,对应约 1200 个 SNV(图 1B)。约 14% 的肿瘤为低突变,而 0.6% 为高度突变。一例高度突变的肿瘤发生在一名非洲血统的患者中。体细胞 SNV 的发生率与插入和缺失的发生率密切相关。广泛的结构变异很常见,普遍存在基因组重排(GR)倒位和染色体间易位(图 1B)。为了定量拷贝数变异 (CNA)的进化过程,他们采用了经过验证的亚克隆重建策略将每位患者的每个事件注释为克隆(主干)或亚克隆(分支)。核基因组中分别有5.6%(主干:2.9%,分支:1.7%)和1.9%(主干:0.01%,分支:0.5%)缺失和增益(图 1C)。
局限性前列腺癌主要受拷贝数变化驱动,特定事件可引发恶性转化,并随后转移扩散。约三分之一的CNA发生在最近的共同祖先(即亚克隆多样化)之前,且表现为克隆性,其中一部分肿瘤显示出亚克隆全基因组重复的迹象(图1D)。由于肿瘤空间异质性以及亚克隆检测的技术限制(纯度、倍性、癌细胞比例和读取深度),这是亚克隆比例的下限:大多数CNA发生在前列腺肿瘤演化的后期。他们使用共识技术创建了亚组,得到了 5 种克隆拷贝数亚型(CCS;CCS1 至 CCS5)和 7 种亚克隆拷贝数亚型(SCS;SCS1 至 SCS7)图1D)。这些亚型重现并扩展了之前使用低分辨率方法和较小队列生成的亚型。克隆和亚克隆 CNA 特征显示出明显的相互关系。克隆和亚克隆 CNA 亚型均与 ISUP GG 紧密相关。
他们利用 GISTIC 数据库鉴定了 31 个复发性克隆和亚克隆 CNA 的驱动区域。大多数 CNA 驱动基因往往出现在癌症进化的早期:31 个中的 21 个优先为克隆性。其余 31 个中的 10 个在克隆期和亚克隆期发生的频率难以区分,且无任何 CNA 驱动基因优先为亚克隆性(图 1D)。这表明,要么大多数鉴定为亚克隆的 CNA 并未为局限性前列腺肿瘤提供选择优势,要么肿瘤在此进化时期所经历的选择压力存在很大的异质性。他们使用了 207 个肿瘤中匹配的 mRNA 丰度数据来鉴定一致的 CNA 和 mRNA 变化,并将这些数据与文献报道相结合,以确定每个复发性 CNA 的假定驱动基因。
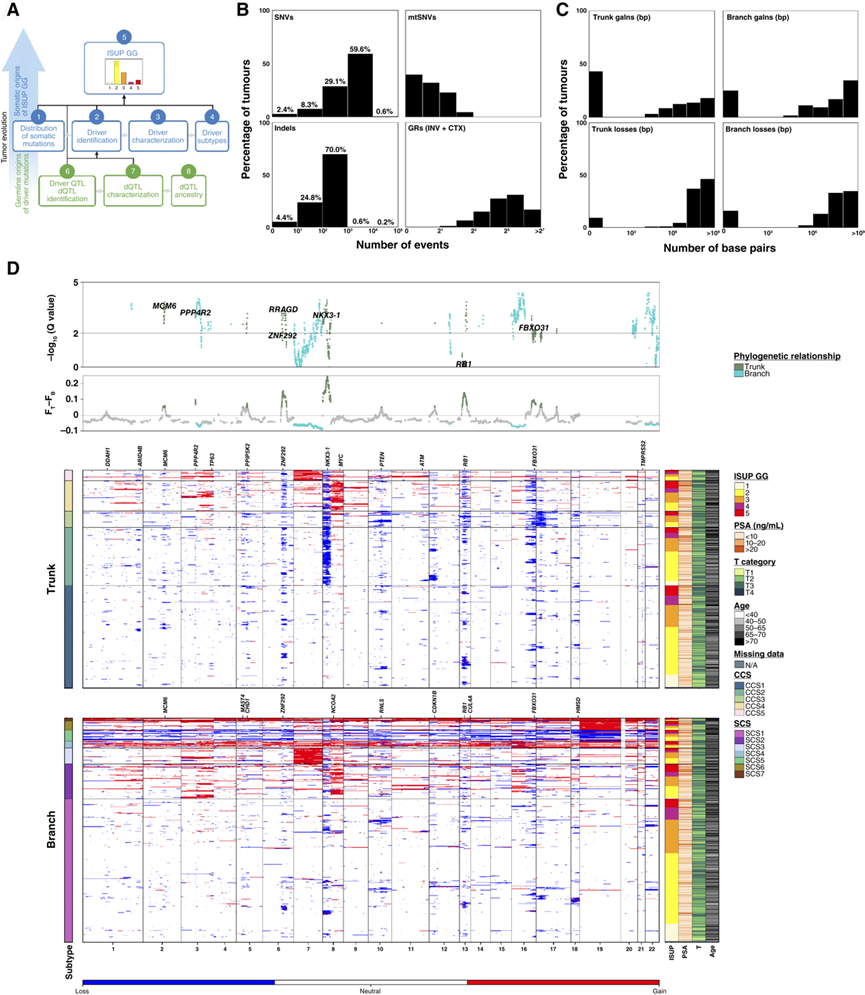
图1. 前列腺肿瘤的突变率。
(A) 本研究中进行的关键分析的示意图。(B) 666对肿瘤-参考全基因组测序(WGS)中SNV、插入/缺失和GR的体细胞突变频率分布。(C) 缺失和增加的体细胞克隆和亚克隆CNA频率(碱基对数)分布。(D) 局限性前列腺癌的克隆和亚克隆CNA概况。
02
局限性前列腺癌的驱动区域
为了补充这 31 个 CNA 驱动区域,他们鉴定了在局部前列腺肿瘤发生和发展过程中受到正选择压力的非 CNA 突变事件。他们应用了 ActiveDriverWGS技术,该技术使用广义线性模型提名复发性突变元素作为候选驱动区域,并根据局部突变特征进行调整。他们鉴定了 39 个受 SNV 和插入/缺失影响的候选驱动区域:13 个蛋白质编码区、24 个非蛋白质编码区和 2 个线粒体区。24 个非蛋白质编码区包括 10 个启动子、10 个增强子、2 个长非编码 RNA (lncRNA)、1 个 miRNA 和 1 个 3' 非翻译区 (UTR)(图S3A)。他们应用 ActiveDriverWGS 鉴定了复发性拷贝中性 GR,鉴定了 110 个驱动区域,包括许多已充分表征的肿瘤抑制因子。为了全面概述局限性前列腺癌的驱动区域,他们还在 1013 个原发性和转移性前列腺癌外显子组的非重叠队列中纳入了 58 个含有蛋白质编码驱动点突变的低频驱动基因。
最终的驱动区域概要包括 31 个 CNA、110 个 GR 和 97 个 SNV/indel。在 20 个患者样本中,多种类型的突变影响了同一区域,导致最终的驱动区域概要包含 223 个:201 个蛋白质编码基因和 22 个非编码区域(图 2)。大多数肿瘤中有 8 个体细胞驱动突变,只有 2% 的肿瘤没有驱动突变,这些肿瘤的治疗前的PSA较低。几乎所有驱动区域(97.3%)都更频繁地受结构变异(CNA 和 GR)的影响,而不是简单的体细胞突变(SNV 和 indel)(图S3B-C)。他们还验证了之前报道的TP53突变与基因组不稳定性之间的关联(图S3D)。
他们汇集了 223 个驱动区域,其中包括一些研究较为深入的突变,例如ETS基因融合(占 51.8%)、线粒体控制区单核苷酸多态性 (SNV)(占 17.9%)以及RB1失活(占 43.2%)和PTEN失活(占 23.4%)。六个先前描述为包含复发性编码单核苷酸多态性 (SNV) 和插入/缺失(IND)的驱动区域,包括ATM、ZNF292和STAB2,与 CNA 和平衡的 GR 驱动区域重叠。多个蛋白质编码基因受到多种突变类型的显著影响,包括TP53和PTEN等关键的抑癌基因。其中包括FOXA1基因座,据报道,该基因座在约 3% 的局限性肿瘤中存在编码点突变,在约 9% 的转移性肿瘤中存在 3′ UTR 插入/缺失,在原发性至晚期疾病中频繁发生结构重排(约 11% 至约 35%)。他们发现了相似比例的非同义 SNV(2.4%)以及复发性编码插入/缺失(3.3%)。这些编码突变伴随FOXA1 3′ UTR 中 SNV 和插入/缺失的显著富集。邻近活性增强子区域 (chr14: 38037521-38073317) 中非编码 SNV 和插入/缺失也显著富集,匹配样本中的 H3K27Ac 染色质免疫沉淀测序 (ChIP-seq)结果证实了这一点。基序分析预测大约一半具有非编码 SNV 的患者会出现转录因子结合基序破坏。FOXA1 基因座中的这些不同类型的突变与FOXA1 mRNA 丰度升高持续相关(图S3E)。14.4% 的患者发生单一FOXA1突变,另有 1.5% 的患者携带两个独立的突变,提示存在双等位基因失活。携带野生型FOXA1的肿瘤易发生鸟嘌呤残基上游四个胞嘧啶 (CpG) 的表观遗传失调,而鸟嘌呤残基之前的胞嘧啶甲基化与FOXA1 mRNA 丰度相关(图S3F),这些数据反映了FOXA1在原发性疾病中失调的多种方式。
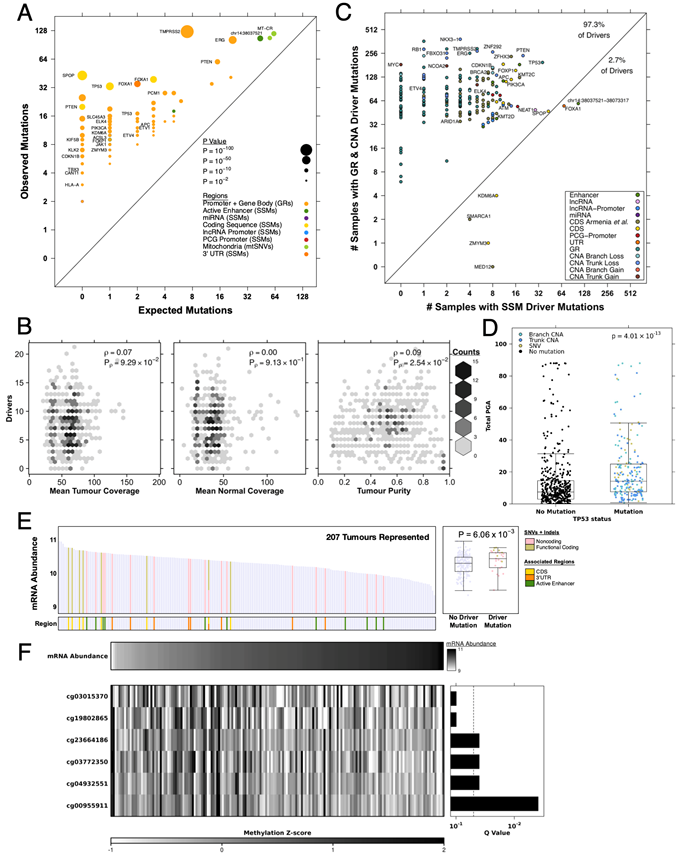
图S3. 驱动突变的性质。
(A)驱动事件的观察和预期突变分析。(B) 每个肿瘤的驱动基因数量与测序覆盖度不相关,仅与肿瘤纯度弱相关。(C) 在666例前列腺癌样本中,驱动区携带CNA或平衡GR的样本数量与携带简单体细胞突变的样本数量比较。(D) TP53突变组与无TP53突变组的总CNA存在差异。(E) 207例肿瘤中FOXA1 mRNA丰度。(F) 在无FOXA1突变的样本中,FOXA1甲基化探针与FOXA1 mRNA丰度相关。
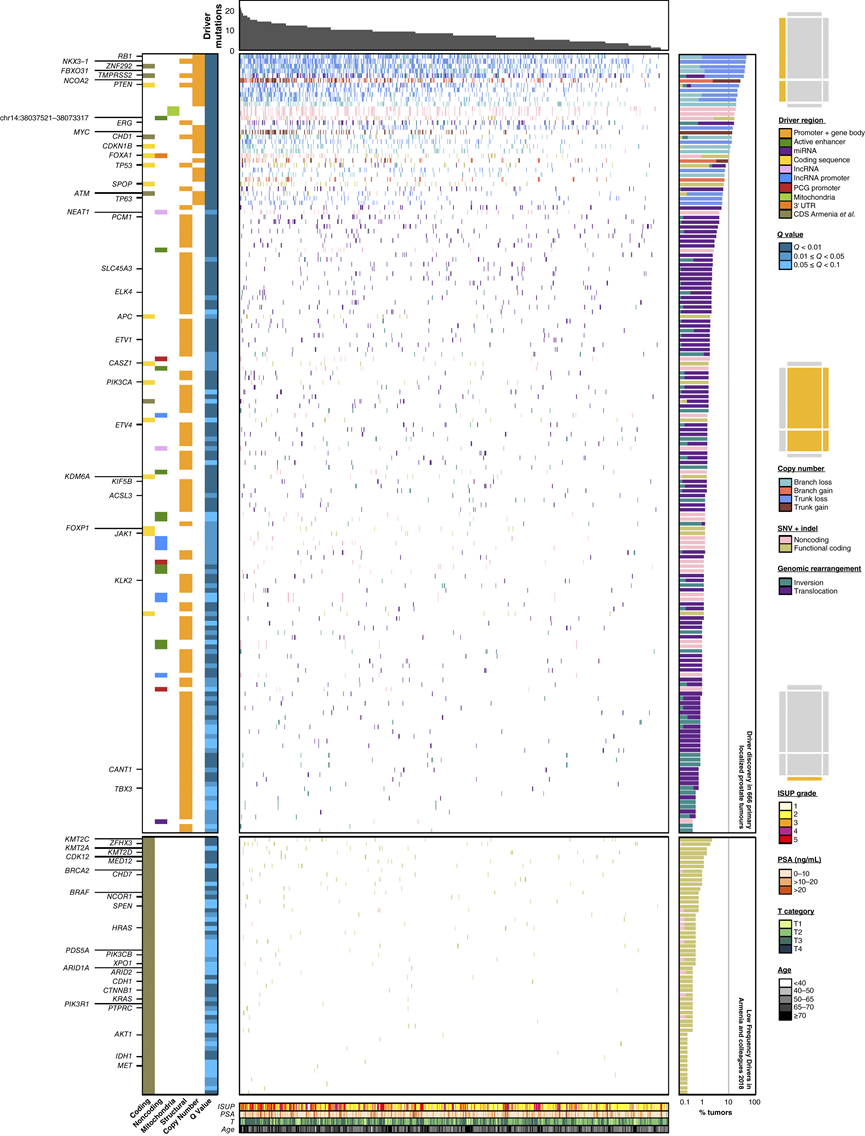
图2. 局限性前列腺癌中的体细胞驱动突变。
03
驱动区域突变影响肿瘤进化
为了确定本研究汇总的 223 个驱动区域是否反映了较少数量的功能组,他们进行了多模态通路分析。50 条通路发生了复发性突变,包括细胞凋亡、有丝分裂和胚胎发育(图 3A)。许多通路因多种突变类型而改变。例如,编码和非编码驱动 SNV 和插入/缺失 [即直接重叠基因或通过染色质环远端关联]优先影响生长和胚胎发育通路,多条通路因许多低频驱动事件而反复改变。
为了确定驱动事件是否影响下游突变过程,他们定量了每种肿瘤的单碱基替换 (SBS) 特征。常见特征包括 SBS8 [同源重组 (HR) 或核苷酸切除修复 (NER) 缺陷] 和 SBS40 (与年龄相关),这与以前的报告一致。在 223 个驱动区域中,有 21 个与突变特征暴露的变化相关(图 3B)。同样,18 个驱动事件与顺式mRNA 变化相关,34 个与影响其他驱动基因的反式变化相关(图 3C)。例如,SPOP突变样本显示CHD1 mRNA 丰度降低,正如预期的那样。
一个驱动区域中的体细胞突变与另一个驱动区域中的转录改变之间的这些反式关联使他们能够定量驱动事件的整体转录组效应。总共有 3318 个转录本与一个或多个驱动因素相关。这些定义了四种失调的m RNA亚型(DMS;DMS1 至 DMS4)(图 3D)。例如,具有SPOP和ZNF292突变的样本显示出相似的转录组失调模式。促进 DMS1 转录表型的驱动因素导致轴突导向调控改变,而促进 DMS2 转录表型的驱动因素则非常广泛地导致基因调控失调(图 3E)。
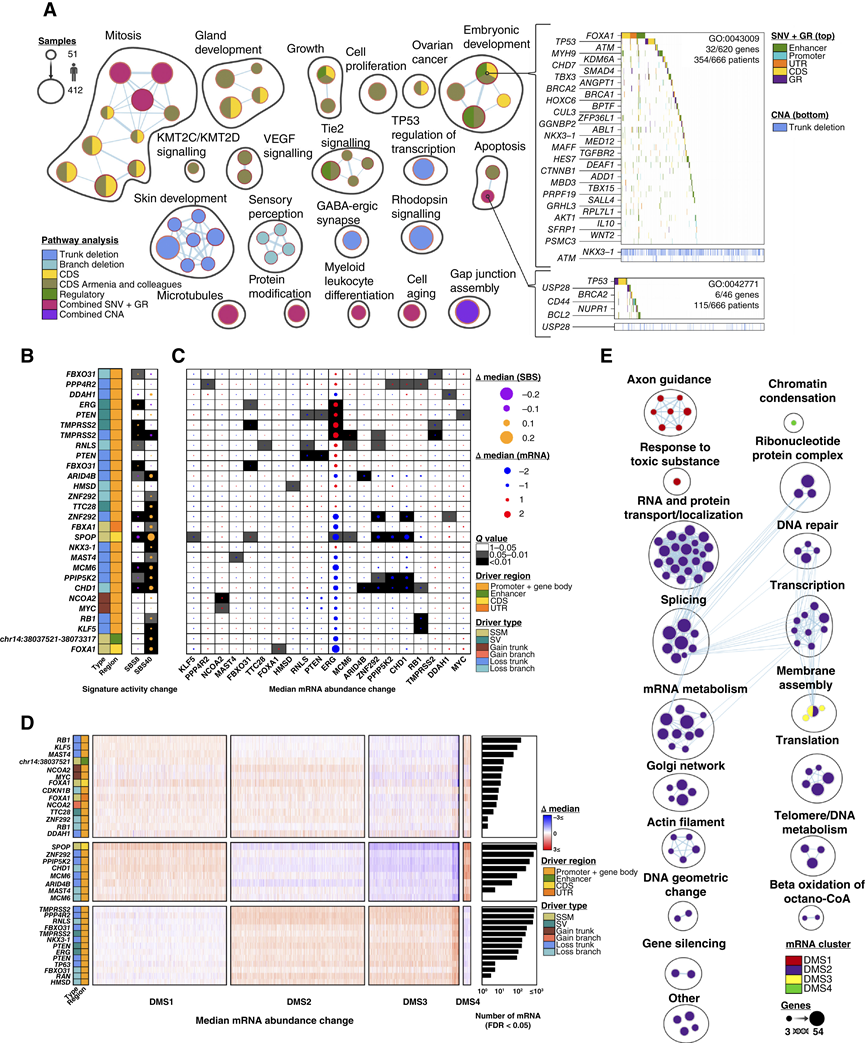
图3. 驱动突变的功能表征。
(A) 网络图表示驱动基因的多模态通路富集分析。热图显示队列中影响导致两种典型通路的基因的突变,而这两种通路受多种突变类型失调。(B) 驱动事件与 SBS 特征之间的关联。(C) 驱动事件与驱动基因 mRNA 丰度之间的关联摘要。(D) 与驱动突变相关的 3318 个失调 mRNA 的一致性聚类。(E) (D)中四种 mRNA 亚型的通路富集分析。
04
局限性前列腺癌的综合分子亚型
由于驱动事件同时发生,孤立地探究每个驱动事件的进化影响无法捕捉前列腺肿瘤基因组的全部复杂性。例如,ETS融合和NKX3-1缺失是两种最常见的驱动突变。两者都发生在肿瘤进化的早期,并且通常具有克隆性。它们在 23.5% 的肿瘤中同时发生。NKX3-1缺失与几乎所有类型的体细胞突变负担增加相关,而ETS融合与拷贝数丢失增加相关(图 4A)。缺乏任一驱动因素突变的肿瘤表现出 SNV 突变率显著增加且无法解释的变异性(图 4B)。八个驱动突变与ETS状态、NKX3-1缺失或两者相关,包括MYC获得和PTEN丢失(图 4C)。
在至少 15 名患者中存在的 61 个驱动事件中,82 对共现,17 对互斥(图 4D)。一组共现驱动因素包括SPOP SNV 以及ZNF292和MCM6的克隆缺失。重现了CHD1、SPOP之间众所周知的关联以及它们与TMPRSS2–ERG基因融合的互斥性。克隆 CNA 驱动因素大多与其他克隆 CNA 驱动因素、SNV/indel 驱动因素(例如FOXA1和SPOP)和 GR 驱动因素(TMPRSS2-ERG)共现。相比之下,MYC克隆增加与四个亚克隆驱动基因同时发生:CUL4A增加、FGF9增加、ZNF292缺失和PPIP5K2缺失。
双等位基因肿瘤抑制基因失活并不常见。同一基因的 CNA 驱动基因的克隆和亚克隆畸变经常相互排斥,这表明现代 CNA 亚克隆检测算法要么针对纯合缺失进行选择,要么存在偏差(图 4D)。同样,没有患者同时存在PTEN的 SNV/插入/缺失和 CNA/GR,这支持单等位基因PTEN缺失足以加速肿瘤发生的假设。系统地评估相互排斥的关联至少需要 1063 个肿瘤才能识别单个肿瘤抑制基因。因此,他们可能已经确定了大多数共现关联,但许多相互排斥的驱动基因对仍然未知。
鉴于突变密度和特定驱动因素彼此之间以及与临床预后特征之间的密切关联,他们首次寻求开发涵盖局限性前列腺癌所有突变类型和等级的整合基因组亚型:之前的所有基因组亚型都仅使用了一部分突变类型。他们生成了七种整合的分子亚型(IMS;IMS1 至 IMS7)(图 4E)。IMS1 是一种突变稳定亚型,占所有患者的 9%(60/647)。该亚型肿瘤的驱动突变数(中位数:2)和所有类型中的总突变数最低,包括所有 13 名未发现驱动突变的患者。大多数 IMS2 肿瘤携带NKX3-1缺失,其他驱动突变很少,而 IMS3 至 IMS7 则具有更多的驱动突变。IMS3 肿瘤倾向于显示 ETS 融合。大多数 IMS4 肿瘤有RB1缺失。IMS5 肿瘤亚克隆拷贝数缺失增多。IMS6 倾向于显示ZNF292缺失以及MCM6和SPOP突变丢失。IMS7 肿瘤以MYC扩增为特征并且 ISUP 等级更高(图 4F)。亚型不受患者年龄、肿瘤范围或治疗前 PSA 的影响,但与治疗后复发有关(,与风险分层方案中等级的临床优势一致。
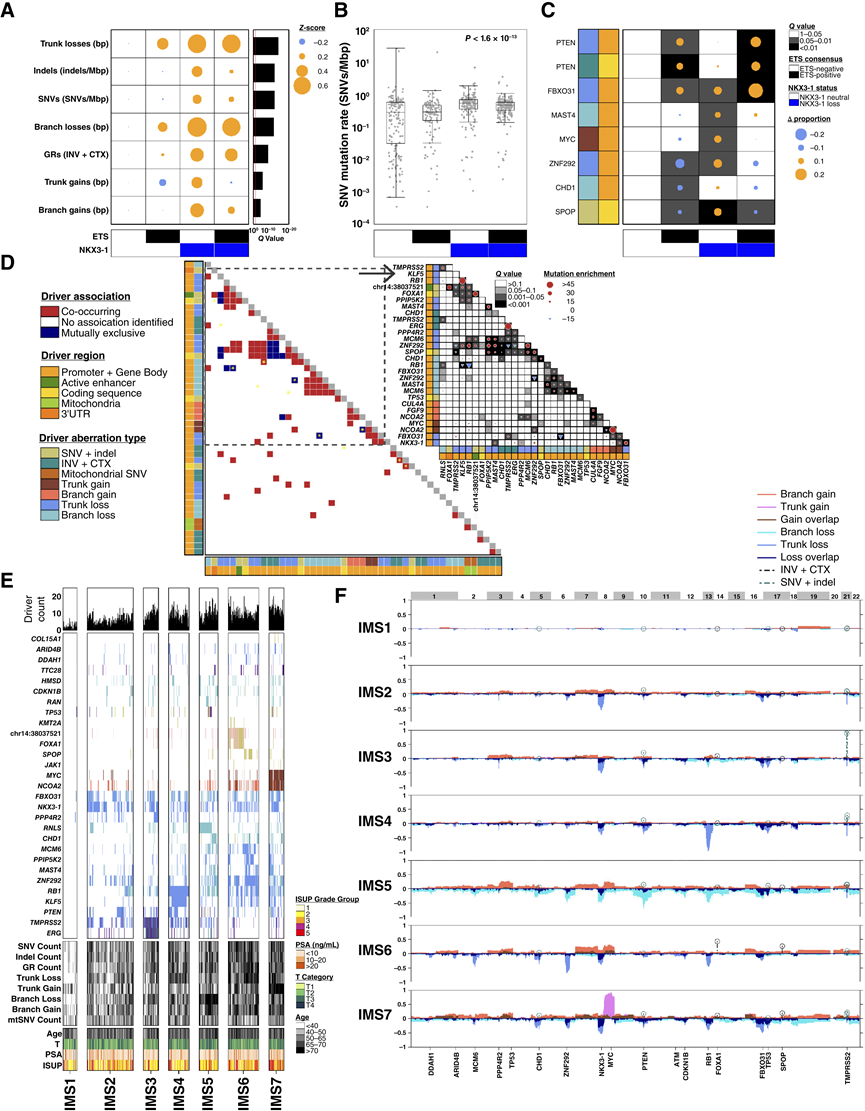
图4. 局限性前列腺癌的突变亚型。
(A) 突变密度(行)因 ETS 融合和NKX3-1 CNA 状态(列)而异。(B) 比较患者除以 ETS 融合和NKX3-1 CNA 状态后的SNV 突变率。(C) 使用广义线性模型,确定了 8 个驱动突变,这些突变的频率因 ETS 融合和/或NKX3-1 CNA 状态而异。(D) 666 例局限性前列腺肿瘤中驱动区域对的共现和关联。(E) 驱动区域聚类识别出七种患者亚型:IMS1–IMS7。柱状图表示患者。底部行显示临床特征,第二行显示突变密度,第三行显示不同亚型之间频率不同的驱动突变。顶部条形图显示了每位患者的突变驱动区域数量。(F) 亚型概况摘要,显示了亚型中存在特定异常的患者比例。
05
临床预后特征的分子相关性
突变亚型与肿瘤分级之间的这种紧密关联促使他们探索不同 ISUP GG 肿瘤之间的突变差异。控制肿瘤和正常测序覆盖度,高级别肿瘤的特点是突变负担大幅增加(图 5A)。克隆和亚克隆 CNA 以及 SNV 均是如此:GG 1 肿瘤中的 SNV 中位数为 803 个,而GG 5 中为 1401 个,重现了先前的研究结果。
较高级别的肿瘤具有更多的驱动突变(图 5B)。在 223 个驱动区域中,有 14 个与 ISUP GG 单变量相关,最显著的是MYC增益(图 5C)。大多数与级别相关的驱动突变优先在肿瘤演变的早期以克隆方式发生:与 ISUP GG 相关的 14 个驱动区域中的 9/14 是 CNA,而 8/9 优先以克隆方式发生。驱动因素表现出四大类与级别的关联,他们将其称为驱动聚类 MG1 至 MG4。MG1 与级别无关,而 MG2 和 MG3 与级别关联较弱,这与基因组不稳定性上升相一致。 MG4 包含与等级密切相关的驱动区域:MG4 中的典型基因在 ISUP GG 1 中发生突变的概率为 28.3%,但在 ISUP GG 5 中发生突变的概率为 49.5%。ISUP GG 和 IMS 之间也存在关联。
他们将这些分析扩展到局限性前列腺肿瘤的其他临床预后特征:诊断年龄、治疗前血清 PSA 浓度和肿瘤范围(T 分类)。对于每一项,他们都确定了与突变密度和特定驱动突变的关联。例如,年龄与 SNV、插入/缺失和亚克隆 CNA 负担增加有关,但有趣的是,与克隆 CNA 无关(图 5D-E)。六个驱动突变与诊断年龄有统计学相关性,十四个与血清 PSA 丰度有统计学相关性,七个与肿瘤范围有统计学相关性。与不同临床特征相关的基因几乎没有重叠:没有基因与所有四种基因相关,这与它们独立的预后能力一致(图5F)。这些数据与诱变领域出现的所有级别的疾病一致,早期克隆驱动突变决定了低级别和高级别的发展轨迹。
他们重点研究了与临床预后特征相关且在至少 1% 的患者中发生突变的 35 个基因。其中六个与结果相关(图 5G),五个在调整临床预后特征后仍然显著。在这五个基因中,MYC和NCOA2增益经常同时出现,因为它们都在 8q 染色体上,而所有其他对均未同时出现。突变时机是决定突变对患者预后影响的主要因素,越早发生的突变通常影响越大。克隆性MYC增益比亚克隆性MYC增益具有更强的预后预测能力(图5H)。同样,PTEN的克隆性缺失而非亚克隆性缺失与更差的预后相关。因此,大多数驱动突变发生在前列腺癌发展的早期,可能与疾病的发生有关。这些早期发生的驱动突变中的一小部分也与特定的临床预后特征和发展为致命性疾病相关。
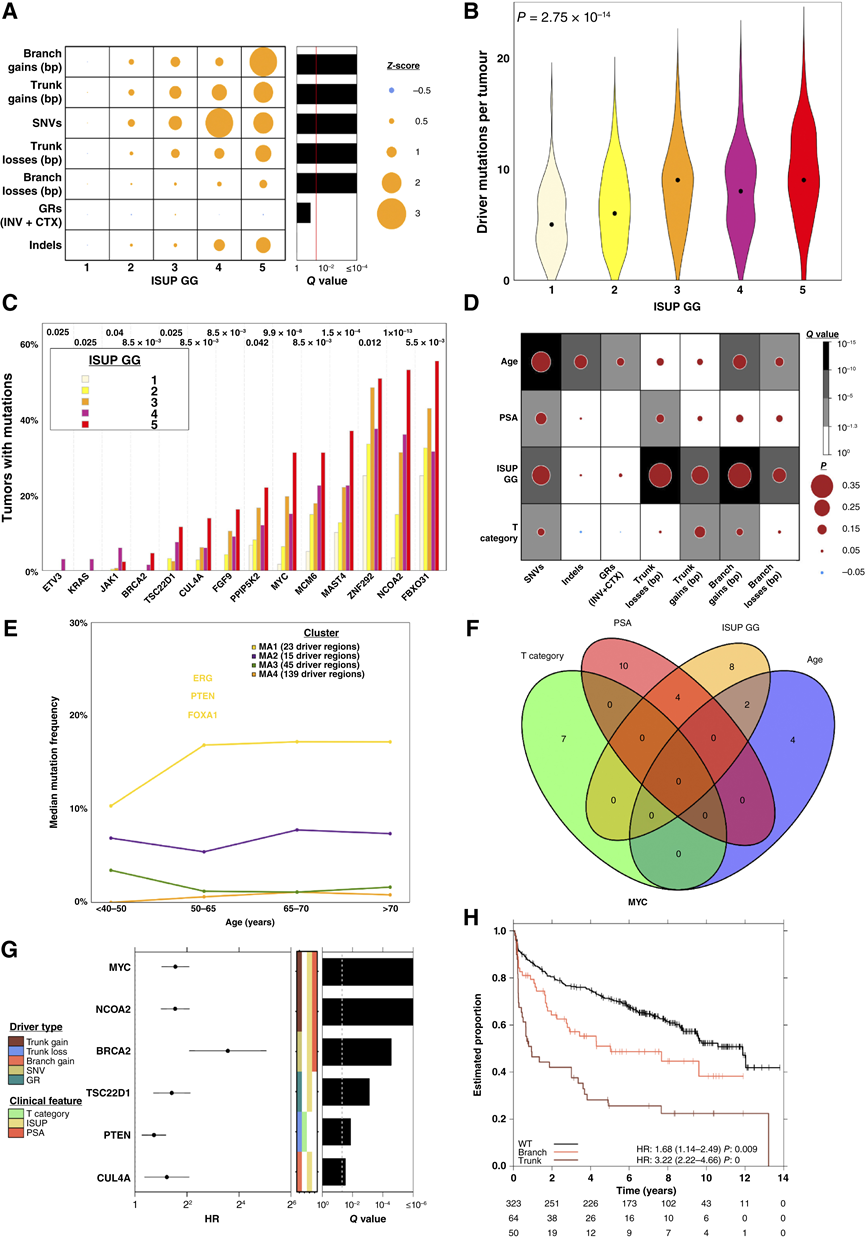
图5. 前列腺癌分级的突变特征。
(A) 使用肿瘤和正常测序覆盖率作为协变量,拟合线性模型,将每个突变密度测量值与 ISUP GG 关联起来。(B) 每个 ISUP GG 中每个肿瘤的驱动突变数量分布,每个 GG 的中位数用黑点表示。(C) 突变频率与 ISUP GG 单变量相关的基因,按 ISUP GG 5 肿瘤中突变样本的百分比排序。(D) 临床协变量与基因组不稳定性测量值之间的双侧 Spearman 相关性,点的大小表示相关性大小,背景颜色表示统计学显著性. (E)共识聚类确定了四组在不同年龄段具有相似变化模式的基因。(F) 与临床特征统计相关的驱动基因的维恩图。(G) Cox 比例风险模型适合与临床特征相关的驱动区域。(H) MYC克隆和亚克隆增益与生化复发相关。
06
体细胞突变驱动因素的种系相关性
鉴于前列腺癌是遗传性最强的实体瘤类型之一,他们接下来试图确定特定的驱动突变是否与特定的种系 SNP 相关,并将这些关系称为驱动数量性状基因位点 (dQTL)。全面开展全基因组关联研究 (GWAS) 需要数千名肿瘤患者进行WGS,但目前尚无此方法,因此他们利用四种针对性策略来富集候选 dQTL(图 6A)。
他们首先考虑了147个风险等位基因,重点关注次要等位基因频率(MAF)> 0.05的134个(图6A)。其中6个与一个或多个躯体驱动因素相关(图6B)。在根据ISUP GG、T分类和PSA调整指数事件偏向后,所有6个dQTL仍然显著。
其次,他们评估了体细胞事件边界±500 kbp范围内的常见SNP(MAF > 0.05),将17个体细胞驱动因素分别与1332至11618个生殖细胞SNP进行比较。在控制群体结构和体细胞突变负荷后,在11个单倍型区块中鉴定出20个局部dQTL,涉及5个驱动因素(图6B)。
第三,他们根据DNA二级结构定义了与体细胞事件的接近度(图6A)。基于前列腺细胞系中的RNA聚合酶II (RNAPII) ChIA-PET和 RAD21 ChIA-PET 定义了空间局部dQTL。如果线性局部边界之外的区域与至少两种细胞系中的事件区域发生相互作用,则将其视为局部dQTL。在此步骤中,评估了17个体细胞驱动因素与7至101个SNP的关联性。他们发现了两个与RB1克隆(主干)丢失相关的dQTL (图6B)。
第四,他们考虑了接近度,其定义是通过在前列腺癌细胞中进行 HiChIP H3K27ac 分析所识别的相互作用增强子(图 6A)。他们确定了锚对,其中一个锚位于驱动区域内,另一个位于驱动区域外。评估了 17 个体细胞驱动因素与 0 至 1059 个 SNP 的关联。他们确定了 11 个 dQTL,涉及七个单倍型区块和三个体细胞驱动因素(图 6B)。
本研究的四种 dQTL 发现策略鉴定了 26 个标签 dQTL,涉及 25 个独特基因座(图 6B)。其中,16 个在 552 名患者的复制队列中显示出一致的效应大小,大部分与外显子组测序数据有关(图 6C)。其中包括四个非常强的效应:rs11203152 缺失TMPRSS2,rs141393446缺失ZNF292 ,以及 rs848047 和 rs848048 均在FOXA1的 3′ UTR 中存在 SNV(图 6D-G)。
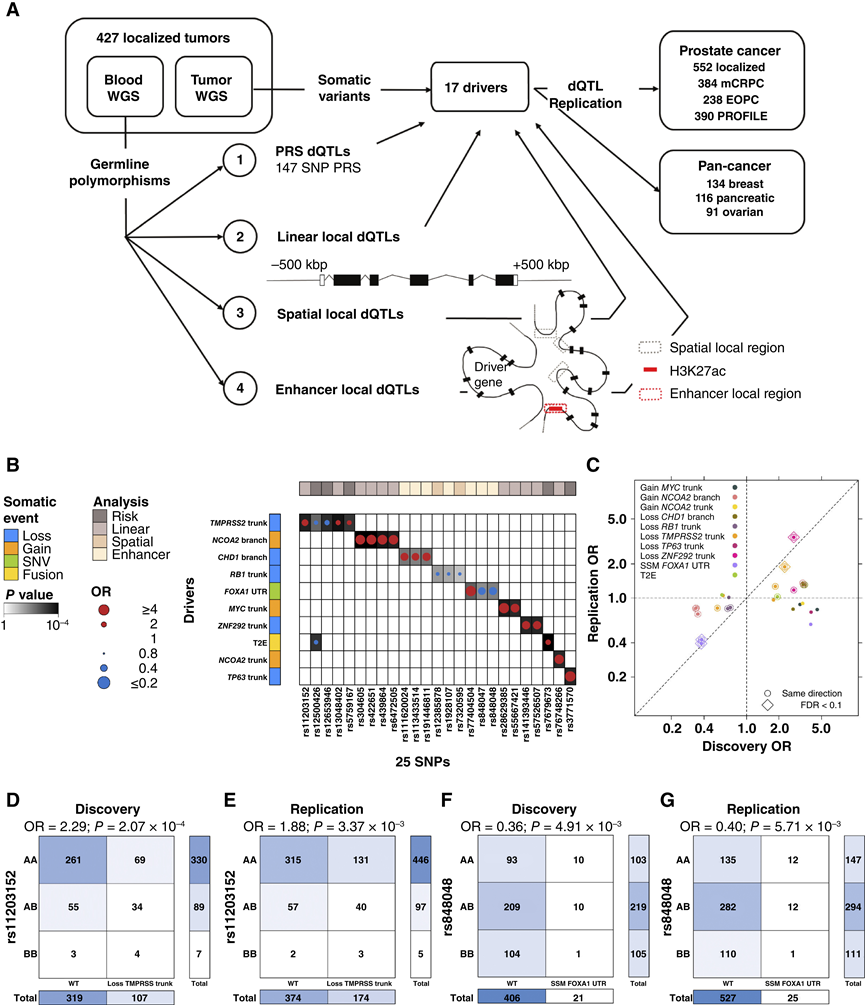
图6. dQTL 偏向体细胞突变景观。
(A) dQTL 检测示意图。(B) 涉及 25 个独特变异的 26 个 dQTL 的摘要。点的大小和颜色表示 SNP 和体细胞驱动基因之间 OR 的大小和方向。(C) 发现队列与复制队列中标签 dQTL 的 OR 比较。(D-E) rs11203152 与发现队列和复制队列中TMPRSS2克隆缺失关联的列联表。(F-G) rs848048 与发现队列和复制队列中FOXA1 3′ UTR中的 SNV 关联的列联表。
07
dQTL 在不同进展阶段和癌症类型中复制
他们重点关注复制队列中具有一致 OR 的 16 个 dQTL,并在候选分析中针对所有 17 个体细胞驱动因素筛选了每个标签 SNP。这确定了 9 个候选远端 dQTL(图 7A),其中 7 个重复,总共确认 23 个 dQTL。
为了确定 dQTL 是否影响多种癌症,他们测试了 20 个标签 dQTL,这些标签 dQTL 影响≥5% 的卵巢癌、乳腺癌或胰腺癌中突变的驱动因素,这些癌症类型具有足够的样本量(n > 91)、已知的遗传性和共有的驱动事件。在其他癌症类型中,14 / 20 dQTL 的效应大小一致。最后,他们对1991例欧洲血统前列腺肿瘤进行了Meta分析,其中包括本研究的发现和复制队列、238例早发性前列腺癌 (EOPC) 肿瘤、384例转移性肿瘤以及来自 PROFILE 队列的 91例转移性前列腺肿瘤和 299例局限性前列腺肿瘤。在发现和复制队列中表现出一致效应的 23个 dQTL 中,11个是可复制的 dQTL(图 7B)。因此,dQTL 可以推广到前列腺癌的各个分期以及其他癌症类型。
dQTL 的发现需要匹配的血液和肿瘤组织图谱。尽管使用了目前规模最大的全基因组测序前列腺癌队列,但其统计功效仍低于现代 GWAS 队列。对于常见的体细胞驱动因素(频率为 5% 至 20%)。他们还是鉴定出了 35 个 dQTL,涉及 11 个体细胞驱动因素和 27 个 SNP(图 7A)。他们推断至少还有 314 个 dQTL 有待发现,其效应大小与本研究中鉴定的相似。类似于许多早期 GWAS 分析的亚阈值分析支持大量未识别的前列腺 dQTL 的观点。
为了支持 dQTL 的功能性后果,他们定量了它们对肿瘤基因表达的影响。肿瘤甲基化失调是种系基因组影响癌症风险的一种机制。他们利用了 226 名发现队列患者、412 名复制队列患者和 47个组织学上非恶性前列腺组织基化组数据。他们鉴定并验证了 110 个甲基化数量性状位点 (meQTL),涉及 8 个 dQTL。这显著高于偶然预期。三个 SNP 与肿瘤特异性 meQTL 有关:它们与肿瘤的甲基化变化有关,但与正常前列腺无关。
为了探索 dQTL 是否与其他表观基因组特征相关,他们研究了原发性前列腺肿瘤中组蛋白修饰的 H3K27ac、H3K27me3和 H3K4me3以及雄激素受体(AR)结合情况。在代表全部独特变异的 16 个标签 dQTL 中,有 10 个与活性调控区重叠:6 个 dQTL 与 H3K27ac 位点重叠(2-89 例患者),其中 5 个也与 H3K4me3(1-47 例患者)位点重叠。5 个 dQTL 与 H3K27me3 重叠,其中一个与其他患者的 H3K27ac 位点重叠,提示存在二价染色质。他们在第二组 48 个原发性前列腺癌肿瘤中重复了这些结果,通过 ChIP-seq 对 H3K27ac、H3K4me2、H3K4me3、FOXA1 和 HOXB13 进行了分析。H3K27ac 修饰位点的五个 dQTL 中有两个在肿瘤组织中表现出等位基因失衡,而在正常组织中没有,表明存在等位基因特异性调控。在 16 个 dQTL 标签 SNP 中,有 13 个与五种前列腺细胞系中的活性调控区和主要转录因子结合位点重叠(图7C)。
为了阐明 dQTL 的机制,他们专注于 rs11203152,因为它与TMPRSS2的缺失有关,而 TMPRSS2 的缺失与 AR 结合有关(图 6D-E)。rs11203152 与由 RNAPII、RAD21、AR 和 ERG 锚定的多个染色质环路位点非常接近(图 7D)。为了定量 rs11203152 附近调节染色质环的富集,他们检测了 rs11203152 1 Mbp 范围内的锚定数量是否多于偶然预期。 RAD21、RNAPII、AR 和 ERG 中的锚点均在 rs11203152 周围富集(图 7E),这表明该种系 SNP 可能与 AR 调控相互作用,从而促进TMPRSS2的缺失。
只有两个 dQTL 标签 SNP 作为其相关体细胞驱动基因的表达数量性状基因位点 (eQTL) 发挥作用:一个作用于RB1 mRNA ,一个作用于TMPRSS2(图 7C)。这些 RNA 变化传播到蛋白质丰度。dQTL 通常不会影响近端基因表达,定义为±500 kbp,仅针对 rs12653946-IRX4鉴定了一个额外的 eQTL ,验证了之前的研究。同样,只有两个 dQTL 影响远端基因表达。最后,鉴于许多体细胞突变以及IMS突变与前列腺癌侵袭性相关,他们评估了dQTL是否可以预测特定的临床特征。一个dQTL与生化复发相关。四个dQTL与诊断时的ISUP GG相关。一个dQTL与前列腺癌诊断风险相关(图7C)。总体而言,他们发现 dQTL 负荷与生化复发和 ISUP GG 之间存在正相关性,虽然相关性不显著,这表明 dQTL 可能是进一步完善预后 PRS 的有价值的候选对象。
已充分证实,遗传祖先与前列腺癌体细胞景观的特定特征相关,但尚不清楚特定的种系 SNP 是否对这些差异的很大一部分做出了贡献。他们接下来关注与两种具有强祖先关联的突变相关的 SNP:T2E 和FOXA1。T2E 基因融合在非洲和东亚血统的个体中较少发生。rs11203152 dQTL 与发现队列和复制队列中的TMPRSS2缺失风险增加相关。与这些祖先趋势一致,与欧洲人相比,非洲和东亚人群中该 SNP 的 VAF 明显较低。rs11203152 与TMPRSS2缺失的关联在 115 名非洲血统的男性中显示出类似的效应。
与欧洲血统的男性相比,非洲血统的男性更容易出现FOXA1 SNV,而在东亚血统的男性中,其他血统中未发现的 SNV 热点却很常见。rs848048 dQTL 标签 SNP 与FOXA1 3′ UTR 中 SNV 的发生呈负相关(图 6B)。与这些血统差异一致,该标签 SNP 在非洲人群中的 VAF 明显低于欧洲或亚洲人群,因此可能解释了在没有这种保护性种系 SNP 的情况下FOXA1 SNV 负担更高的原因。非洲个体的等位基因分布与欧洲个体存在显著差异,且这种关联性在非洲人群中并未得到验证,这支持生殖细胞系在祖先相关体细胞差异中的作用。T2E基因的16.7%至31.4%的祖先差异和FOXA1基因的0.9%至67.3%的祖先差异与单个dQTL有关(图7F-G)。
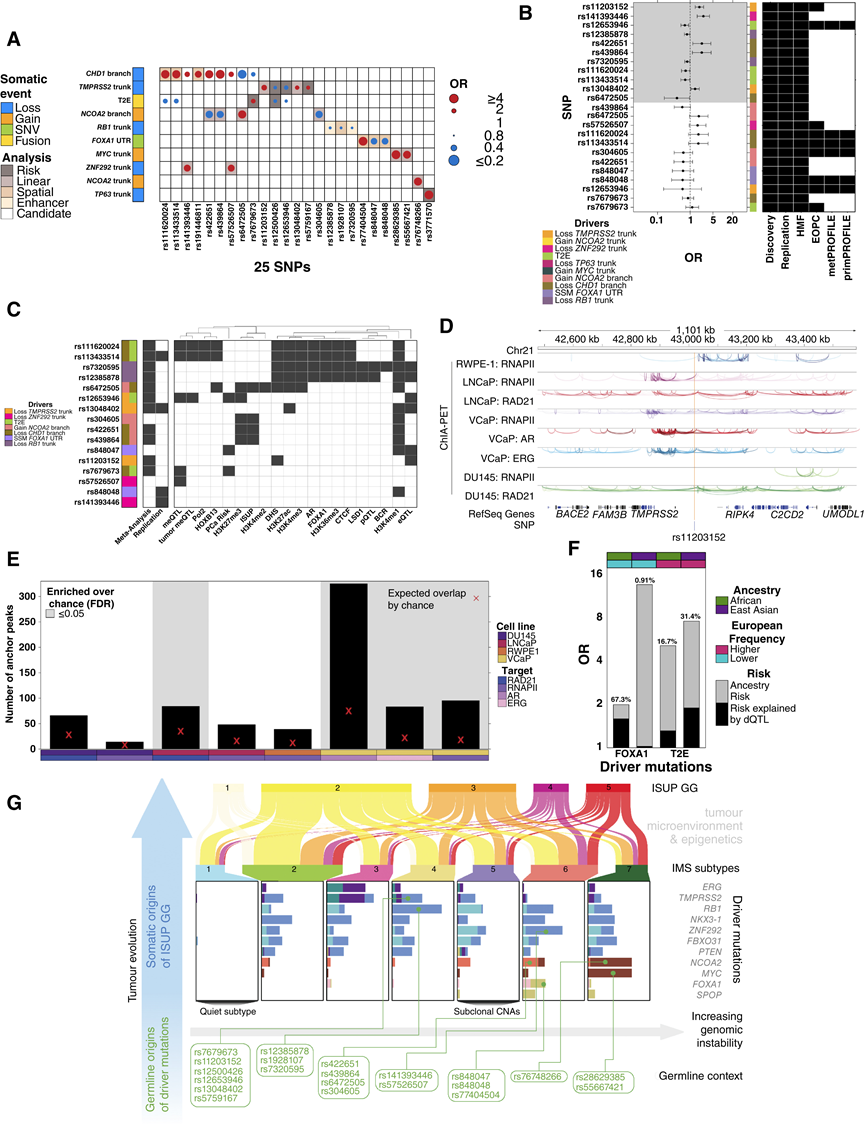
图7. dQTL 的特征。
(A) 涉及 25 个独特 SNP 的 35 个 dQTL 的总结。(B) 1991 例前列腺肿瘤中 dQTL 关联的 OR 和 95% 置信区间的森林图。(C) dQTL 的分子和临床特征总结。(D) rs11203152 位于调控致密区内。轨迹显示 RWPE-1、LNCaP、VCaP 或 DU145 细胞系中由 RNAPII、RAD21、AR 或 ERG 锚定的染色质环路。(E) LNCaP 和 VCaP 细胞系中的染色质环路数量高于偶然预期。(F) dQTL 可能解释不同祖先体细胞突变频率的差异。(G) 原发性前列腺癌演变为 ISUP GG 的示意图。
+ + + + + + + + + + +
结 论
本研究分析了666个前列腺肿瘤全基因组,鉴定出223个反复突变的驱动区域,其中大多数影响着下游的突变过程和基因表达。本研究鉴定并验证了导致肿瘤易获得特定体细胞驱动突变的个体种系变异:这些变异解释了疾病表现的异质性和祖先差异。高级别肿瘤具有低级别肿瘤中驱动基因的超集,包括BRCA2和MYC突变频率的增加。与级别相关的驱动突变发生在肿瘤演化的早期,它们的早期发生强烈预示着癌症的复发和转移。本研究数据表明,高级别和低级别前列腺肿瘤均来自共同的癌前病变领域,受种系基因组背景和随机突变时间的影响。
+ + + + +



