

 English
English文献解读|Cell Metab(30.9):肠道菌群衍生的脱氧胆酸塑造免疫抑制性肿瘤微环境,并促进乳腺癌进展
✦ +
+
论文ID
原名:Gut microbiota-derived deoxycholic acid shapes an immunosuppressive tumor microenvironment and promotes breast cancer progression
译名:肠道菌群衍生的脱氧胆酸塑造免疫抑制性肿瘤微环境,并促进乳腺癌进展
期刊:Cell Metabolism
影响因子:30.9
发表时间:2026.05.12
DOI号:10.1016/j.cmet.2026.04.011.
背 景
乳腺癌仍然是全球女性最常见的恶性肿瘤,也是导致女性癌症相关死亡的主要原因之一。尽管早期检测和全身治疗取得了显著进展,但转移性疾病和治疗耐药性仍然限制着患者的长期生存,这凸显了迫切需要识别驱动乳腺癌进展的新型生物学机制和治疗靶点。越来越多的证据表明,肠道菌群是宿主代谢、免疫和癌症生物学的关键调节因子。肠道微生物组成和功能的改变与多种恶性肿瘤的发生和发展密切相关,包括结直肠癌、黑色素瘤、肺癌、前列腺癌和乳腺癌。值得注意的是,据报道,乳腺癌是实体瘤中微生物丰度和多样性最高的肿瘤之一,且乳腺癌患者与健康对照组的肿瘤内和肠道菌群谱均存在显著差异。此外,不同的微生物特征与乳腺癌的肿瘤分期、分子亚型和治疗反应相关。
实验设计
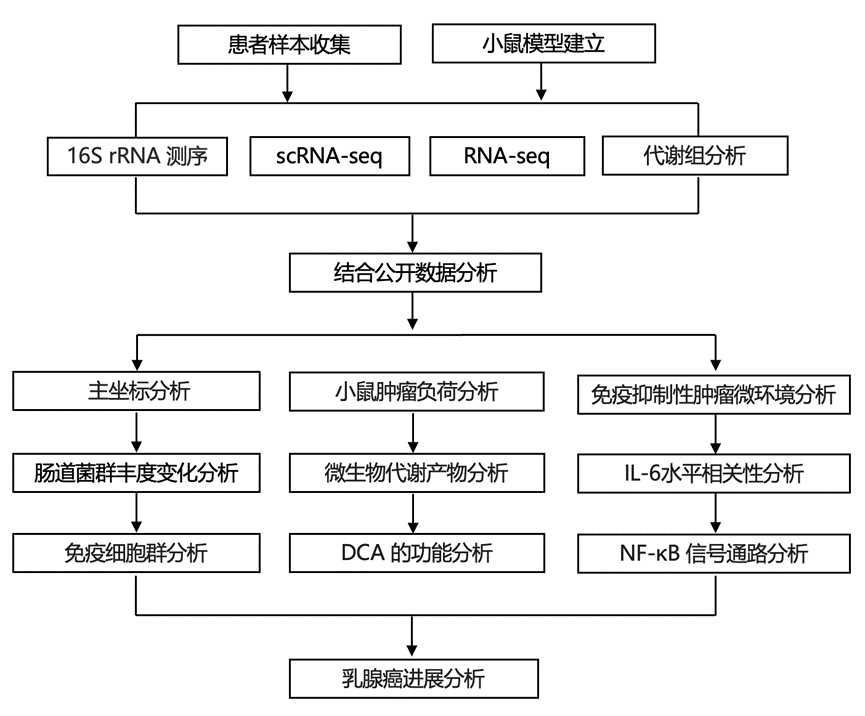
结 果
01

肠道菌群会随着乳腺癌的进展而发生改变
为了确定恶性乳腺癌患者[包括浸润性导管癌 (IDC) 和导管原位癌 (DCIS)]与良性乳腺疾病患者的肠道微生物组成是否存在差异,研究团队进行了 16S rRNA 测序,并基于微生物 β 多样性距离进行了主坐标分析 (PCoA)。该分析显示,IDC 组 ( n=18)、DCIS 组 ( n=7) 和良性乳腺疾病组 (n = 18) 之间存在明显的分离(图1A)。基于 ASV 丰度表生成的花瓣维恩图进一步展示了这些组间共有和特有的 ASV 数量,突出了重叠的微生物成员和组特异性分类单元(图1B)。接下来,他们比较了各亚组间的微生物群落组成,并确定了四个在纲水平上变化最显著的分类单元——Clostridia、γ-变形菌 (Gammaproteobacteria)、拟杆菌 (Bacteroidia) 和Negativicutes(图1C)。为了进一步确定差异富集的分类群,他们进行了线性判别分析效应量(LEfSe),该方法在多个分类水平上鉴定出不同的微生物特征(图S1 A)。在科水平上,厚壁菌门下的毛螺菌科在恶性程度更高的浸润性导管癌(IDC)组中富集,而瘤胃球菌科在良性疾病患者中更为丰富(图1 D)。此外,在恶性肿瘤组(IDC和导管原位癌,DCIS)中,临床分期更晚的患者表现出更高的毛螺菌科相对丰度。
此外,他们建立了小鼠4T1原位乳腺癌模型,并对不同时期(初始、第2周和第4周)采集的粪便样本进行了16S rRNA测序。与临床结果一致,在三个阶段观察到了肠道微生物组成的显著变化。随着肿瘤进展,微生物丰富度(观察到的物种数)下降,而辛普森指数上升,表明群落多样性降低,同时优势类群选择性扩增。进一步的分类学分析显示,在肿瘤进展过程中, Porphyromonadaceae、Prevotellaceae、Rikenellaceae和Bacteroidaceae显著减少,而Lachnospiraceae则显著增加(图1 E)。在属水平上,与乳腺癌进展相关的最显著变化发生在Clostridium_XIVa中。在不同阶段鉴定出的差异性分类群经LEfSe分析后生成系统发育树,该树突状图突显了Firmicute-Clostridia-Clostridiales-Lachnospiraceae- Clostridium_XIVa的显著富集(图1F)。这些结果不仅证实了Lachnospiraceae在恶性乳腺癌微生物群中的富集,而且表明小鼠模型能够有效模拟乳腺癌进展过程中发生的肠道微生物群变化。
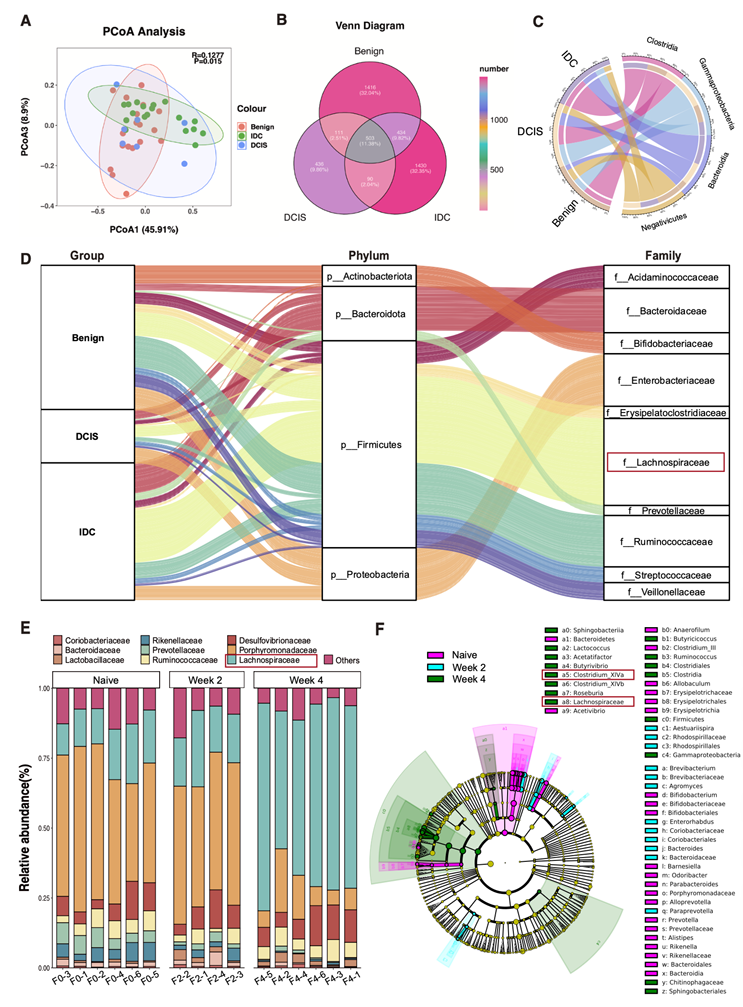
图1. 在乳腺癌的发展过程中,肠道微生物群发生了变化。
(A) 基于加权UniFrac距离的肠道菌群主坐标分析(PCoA)。(B) 图示IDC、DCIS和乳腺良性疾病患者共有和独特的细菌分类群。(C) Circos 图表展示了四个相对丰度较高的细菌类群在 IDC(浸润性导管癌)、DCIS(导管原位癌)和良性乳腺疾病组之间的差异情况,这四个类群的差异在这些组别中最为显著。(D) Sankey图展示了不同类别中在科(右侧)和门(中间)层级上的分类数据随分支宽度的变化情况。(E) 该图展示了在原位 4T1 乳腺癌模型中,未接种组、第 2 周组和第 4 周组肠道微生物群中细菌在属水平上的相对丰度。(F) 原位4T1乳腺癌模型初治、第2周和第4周肠道微生物群差异丰富分类群的进化图。
02
肿瘤相关肠道菌群通过免疫抑制微环境促进乳腺癌进展
接下来,他们利用粪便微生物移植(FMT)技术,从功能上探究了肠道菌群的作用。他们用广谱抗生素清除小鼠体内的内源性菌群四周,随后用来自无肿瘤的正常小鼠(NF)或携带肿瘤的小鼠(TF)的粪便进行两周的菌群重建,之后将4T1细胞原位移植到小鼠体内。值得注意的是,TF组小鼠的肿瘤负荷显著高于NF对照组,这表明TF来源的菌群具有促肿瘤作用。
为了评估E. bolteae的功能影响,他们用E. bolteae(Lach组)或E. coli(E. coli组)对荷瘤4T1小鼠进行口服定植。基于PCR的粪便分析证实,Lach组中E. bolteae显著富集,表明在实验条件下其能够有效定植。Lach组小鼠的肿瘤体积和重量均显著增加(图2 A-B)。为了阐明其潜在机制,他们对Lach组和对照组(Con组)的肿瘤组织进行了单细胞转录组分析(scRNA-seq)。利用基于独特分子标识符的方案,65882个细胞通过了质量控制,并根据典型标记注释为主要群体:肿瘤细胞(Krt18、Epcam和Krt8)、成纤维细胞(Acta2、Col1a1和Col3a1)、内皮细胞(Itga6、Pecam1和Cd93)、粒细胞(Csf3r、S100a8和S100a9)、T 细胞(Cd3d、Cd3e和Ptprc)以及单核细胞/巨噬细胞/树突状细胞(DC)(Cd68、Cd14、Itgam和Ptprc)(图 2C-D)。
为了进一步剖析E. bolteae对肿瘤浸润免疫细胞群的影响,他们对 CD45+细胞进行无监督聚类,得到 16 个免疫亚群:浆细胞样树突状细胞 (pDC)、常规树突状细胞 (cDC)、单核细胞衍生的巨噬细胞、肿瘤相关巨噬细胞 (TAM)、粒细胞来源的髓系抑制细胞(G-MDSC)、Ccl6+巨噬细胞、Nrp2+巨噬细胞、中性粒细胞、Ccl8+巨噬细胞、幼稚 T 细胞、增殖 T 细胞、CD8+ T 细胞、CD4+ T 细胞、B 细胞、γδT/自然杀伤 (NK) 细胞和肥大细胞(图2E)。
在这些细胞群中,G-MDSC在Lach处理的肿瘤中扩增最为显著(图2F)。与中性粒细胞相比,G-MDSC的Clec4e、Il1b和Arg2表达水平更高,这与其已知的免疫抑制表型相符。功能通路分析进一步揭示,Lach定植与Th17分化、抗原加工和呈递、T细胞受体信号传导以及细胞因子-细胞因子受体相互作用通路的激活相关,表明E. bolteae通过扩增免疫抑制性髓系细胞和调节适应性免疫信号传导来促进肿瘤支持的免疫微环境。
既往研究报道,作为一种厌氧菌,E. bolteae对甲硝唑治疗敏感。因此,他们用甲硝唑治疗荷瘤小鼠,观察到肿瘤体积和重量显著降低(图2G-H)。同时,对肿瘤浸润免疫细胞的分析显示,G-MDSC 和 Th17 细胞显著减少(图2I-K)。M-MDSC 基本保持不变,而 CD11b⁺ 髓系细胞减少。相比之下,CD3+和 CD4+ T 细胞的总体频率没有显著变化。为了进一步阐明甲硝唑对肠道菌群的影响,他们在治疗后进行了丰度分析,发现甲硝唑组中Clostridia的丰度显著降低(图2L)。此外,在科的层面上,毛螺菌科表现出显著的减少。
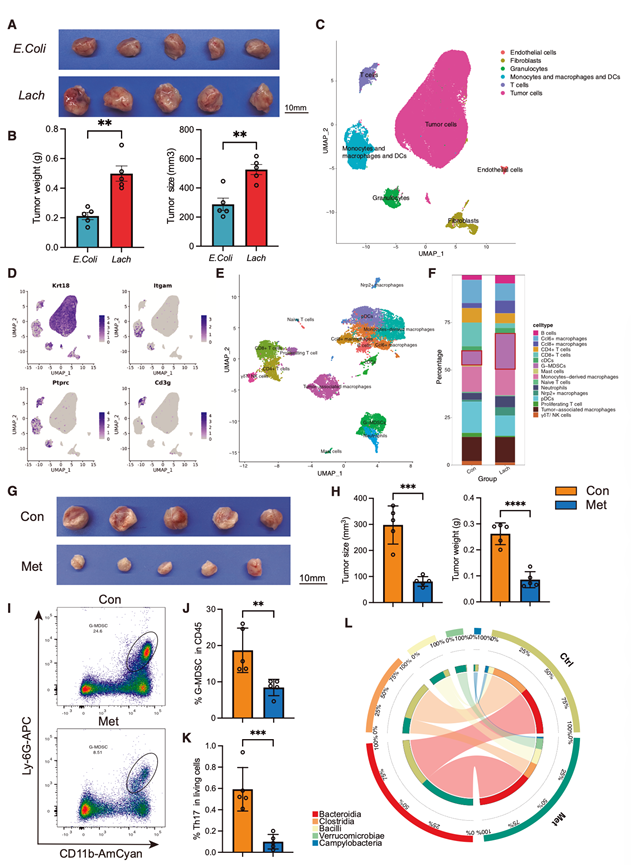
图2. 肿瘤相关的肠道微生物群通过免疫抑制微环境促进乳腺癌的进展。
(A) 4T1 肿瘤荷瘤小鼠(分别接种了肠球菌博特莱菌的 Lach 组或大肠杆菌的 E. coli 组)的肿瘤代表性图像。(B) 对携带 4T1 肿瘤的小鼠在口服接种博特莱氏芽孢杆菌或大肠杆菌后肿瘤负荷的量化结果,以肿瘤重量(左图)和肿瘤大小(右图)的形式呈现。(C) 小鼠肿瘤细胞可视化分析。(D) 展示代表性标记基因在主要细胞类型中表达情况的特征图谱。(E) UMAP图突出肿瘤微环境中的免疫细胞亚群。(F) 堆叠条形图显示Con组和Lach组肿瘤组织中不同细胞类型的比例分布。(G) 甲硝唑治疗4T1荷瘤小鼠(Met,下)与未治疗对照组(Con,上)肿瘤的代表性图像。(H)小鼠的肿瘤负荷量化,显示为肿瘤大小(左)和肿瘤重量(右)。(I) 具有代表性的流式细胞图显示Met和Con小鼠肿瘤浸润的G-MDSC。(J) 对小鼠肿瘤中的CD45+肿瘤浸润免疫细胞群中的 G-MDSC进行流式细胞分析。(K)小鼠肿瘤活细胞中Th17细胞的流式细胞分析。(L)细菌的平均相对丰度。
03
微生物代谢产物DCA在肿瘤进展中起着至关重要的作用
鉴于肠道菌群在促进乳腺癌进展中发挥的因果作用已在上文中得到证实,他们接下来试图阐明其背后的功能机制。越来越多的证据表明,除了直接的微生物-宿主相互作用外,肠道菌群还可以通过微生物来源的代谢产物调节宿主的代谢和免疫通路,从而影响肿瘤生物学。为了进一步评估肠道菌群相关的代谢产物,他们接下来对处于4T1肿瘤不同发展阶段的小鼠粪便样本进行了非靶向代谢组学分析。与无肿瘤对照组(未处理组)相比,荷瘤小鼠(第4周)的粪便代谢组表现出明显的代谢特征(图3A)。荷瘤小鼠中共有140种代谢物富集,而无肿瘤对照组中则有133种代谢物含量较高(图3B)。
为了进一步整合微生物组成与代谢谱,他们结合16S rRNA测序和代谢组学数据进行了整合相关性分析。值得注意的是,胆汁酸相关代谢物——尤其是脱氧胆酸(DCA)——成为富集程度最高且与微生物群相关性最强的候选代谢物之一,凸显了微生物组成变化与代谢组学结果之间的一致性。具体而言,DCA与毛螺菌科的丰度呈最强的正相关性(图3C),表明它可能代表一种关键的微生物群来源介质,将优势肠道菌群与乳腺癌进展联系起来。
DCA的形成是一个多步骤过程,涉及宿主和微生物的酶活性。初级胆汁酸在肝脏中通过细胞色素P450依赖性反应合成,随后进行结合,并分泌到肠道。进入肠道后,进一步转化为次级胆汁酸需要微生物的活性——其中最关键的是表达细菌胆盐水解酶(BSH)的细菌(如Bacteroides、Lactobacillus、Bifidobacterium和Clostridium对胆盐水解的作用,随后是主要由Clostridium和Eubacterium介导的7α-脱羟基化作用。与此相符的是,E. bolteae能够有效地代谢初级胆汁酸并生成DCA。
胆汁酸来源于肠道,其在乳腺组织中的浓度高于血清。为了研究DCA是否与乳腺癌进展相关,他们分析了56例乳腺疾病患者的外周血样本。与良性乳腺疾病患者(n = 31)相比,恶性乳腺癌患者(n = 25)的血清DCA水平显著升高(图3D)。同样,在4T1小鼠模型中也观察到DCA水平随乳腺癌进展而逐渐升高(图3E)。此外,FMT和甲硝唑实验进一步证实了DCA的重要作用,TF组血清DCA水平显著升高,而甲硝唑组血清DCA水平显著降低(图3F-G)。最后,为了评估DCA是否会在肿瘤组织内积累,他们定量分析了NF和TF受体小鼠肿瘤中的DCA水平,以及乳腺癌患者配对的肿瘤(T)和肿瘤邻近正常(TAN)乳腺组织中的DCA水平。与NF组相比,TF组小鼠肿瘤中的DCA水平显著升高(图3H)。同样,在临床样本中,肿瘤组织中的DCA浓度也显著高于相应的TAN组织(图3I)。这些结果表明,DCA不仅在全身范围内升高,而且优先在肿瘤微环境中积累,支持该代谢物具有直接的促肿瘤作用。
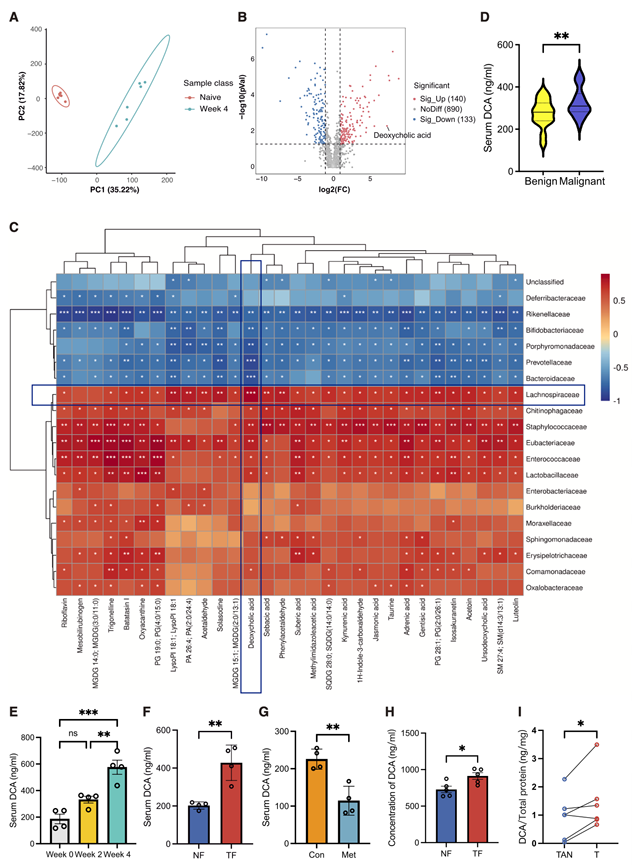
图3. 微生物代谢产物DCA在肿瘤进展中起着至关重要的作用。
(A) 对未患病小鼠和第 4 周肿瘤形成小鼠的粪便代谢物谱进行主成分分析(PCA)。(B) 火山图显示了初代和第4周荷瘤小鼠粪便中微生物代谢物的差异。(C) 前30个微生物类群与荷瘤小鼠代谢产物显著增加的相关性分析。(D) 患有良性乳腺疾病和恶性乳腺癌患者的血清DCA水平。(E) 4T1荷瘤小鼠不同时期血清DCA水平。(F) 4T1荷瘤小鼠接受来自NF或TF小鼠的FMT后血清DCA水平。(G) 用甲硝唑(Met)治疗的 4T1 肿瘤荷瘤小鼠血清中的 DCA 水平与未治疗的对照组(Con)相比。(H) NF-和TF-FMT处理小鼠肿瘤组织中的DCA水平。(I) 乳腺癌患者配对肿瘤(T)和肿瘤邻近正常(TAN)乳腺组织中的DCA水平。
04
DCA通过激活FXR促进乳腺癌的发生
DCA的主要受体是核胆汁酸激活转录因子FXR和G蛋白偶联胆汁酸受体1 (GPBAR1),后者介导膜启动的信号传导。他们利用Kaplan-Meier Plotter在线平台和GEO公共数据库对乳腺癌患者进行了生存分析。法尼醇X受体(FXR)(NR1H4)高表达患者的远处转移无进展生存期(DMFS)显著缩短,而GPBAR1表达对预后无影响。
为了验证这些发现,他们检测了乳腺癌组织样本中FXR的表达。结果与预期一致,在他们纳入的队列中,FXR高表达患者的无远处转移生存期(DMFS)较短(图4A)。此外,与TAN相比,肿瘤组织中FXR的表达显著升高(图4B-C)。他们进一步观察到,与侵袭性较低的管腔型乳腺癌相比,非管腔型乳腺癌(通常恶性程度更高)中FXR的表达更高。这一趋势在乳腺癌细胞系中也得到了证实:与激素受体(HR)阳性的管腔型乳腺癌细胞系MCF-7和T47D相比,代表非管腔型乳腺癌的MDA-MB-231和BT-549细胞中FXR的表达升高。
为了确定FXR是否是DCA诱导肿瘤促进所必需的,他们使用短发夹RNA(shRNA)敲低4T1肿瘤细胞中的FXR,并验证了敲低效率。在测试的shRNA构建体中,sh2-FXR的敲低效率最高,因此用于所有后续的功能分析。实时细胞分析(RTCA)显示,无论是用DCA激活FXR,还是用FXR拮抗剂(ZGS,Guggulsterone E&Z)抑制FXR,亦或是shFXR介导的敲低,均未显著改变体外肿瘤细胞的固有增殖能力。为了探究DCA-FXR轴如何影响体内肿瘤生长,他们采用了多种干预模型。给予E. bolteae定植培养物的条件培养基(LM)可增强G-MDSC和Th17细胞的浸润,并促进乳腺肿瘤的进展。值得注意的是,当小鼠接受源自4T1-shFXR细胞的肿瘤或接受 FXR 抑制剂 ZGS 治疗时,这些效应显著消失(图4D-G)。此外,在4T1-shNC和4T1-shFXR模型中,与大肠杆菌对照组相比,Lach(E. bolteae)组肿瘤内的DCA水平持续升高(图 4H)。此外,在另一个同源乳腺癌小鼠模型(EO771)中,E. bolteae的定植和其条件培养基的给药均能加速肿瘤生长,而甲硝唑治疗则显著抑制肿瘤进展。同样,直接给予 DCA 也重现了这种表型。DCA 暴露伴随着 G-MDSC 和 Th17 细胞在肿瘤内的浸润增加以及免疫抑制性肿瘤微环境的建立。重要的是,无论是使用 ZGS 进行 FXR 的药理学抑制,还是在肿瘤细胞中特异性敲低 FXR,都能有效消除 DCA 诱导的免疫抑制变化并抑制肿瘤进展(图4I-K)。另一方面,使用 SBI-115(一种 GPBAR1 抑制剂)也能部分抵消 DCA 的促肿瘤作用。
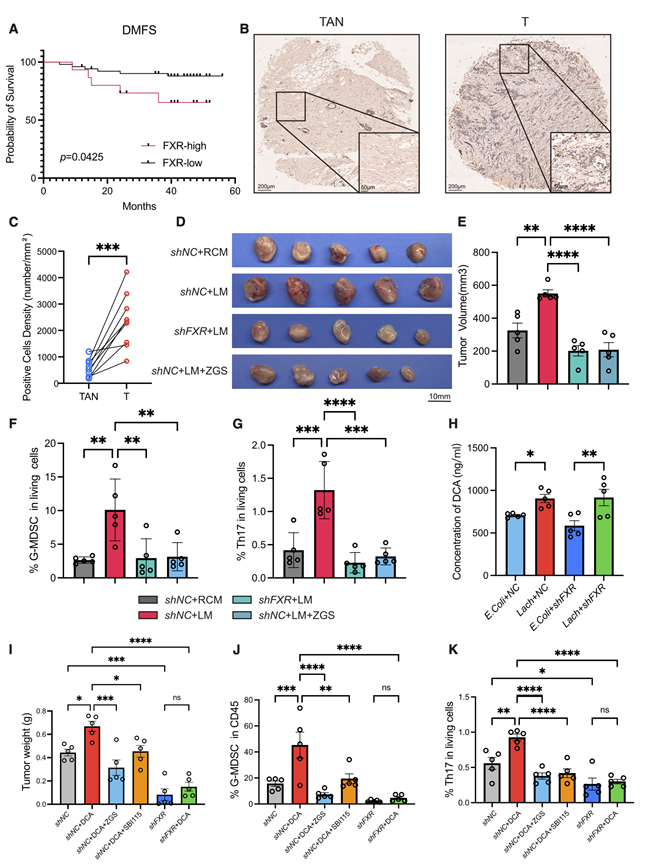
图4. DCA通过激活FXR促进乳腺癌。
(A) 通过NR1H4 (FXR)表达分层的乳腺癌患者的远端无转移生存期(DMFS)的Kaplan-Meier分析。(B) 配对乳腺癌肿瘤组织(T)和肿瘤邻近正常组织(TAN)中FXR的代表性免疫组化(IHC)染色图像。(C) 乳腺癌患者配对T和TAN中FXR阳性细胞密度的统计分析。(D-E) 肿瘤分析。(F-G) 流式细胞分析各治疗组肿瘤浸润细胞中G-MDSC和Th17细胞。(H) 荷瘤小鼠肿瘤组织中DCA水平。(I) DCA补充饮食处理小鼠肿瘤重量。(J) 流式细胞分析各治疗组肿瘤中CD45肿瘤浸润免疫细胞中G-MDSC的表达。(K) 流式细胞分析各治疗组肿瘤浸润活细胞中Th17细胞。
05
IL-6水平升高与DCA介导的肿瘤免疫抑制微环境相关
既往研究表明,DCA可激活食管细胞中的IL-6/STAT3信号通路,从而促进其向食管癌癌前病变转化。为了阐明DCA促进肿瘤进展的机制,他们对对照组和DCA处理组小鼠的肿瘤组织进行了转录组分析(RNA-seq)。DCA补充诱导了显著的转录重塑(图5A)。上调基因的GO富集分析显示,其在与“细胞因子产生的正调控”、“髓系白细胞迁移”、“IL-6产生”和“粒细胞趋化性”相关的生物学过程(BP)中显著富集(图5B)。与此一致的是,细胞因子芯片分析表明,在4T1肿瘤进展过程中,血清IL-6水平显著升高(图5C)。
鉴于 IL-6 在塑造乳腺癌免疫微环境方面发挥着重要作用,接下来他们评估了 DCA 是否直接影响肿瘤细胞的 IL-6 产生。体外实验表明,DCA处理呈剂量依赖性地增加肿瘤细胞IL-6的分泌,而FXR敲低则显著抑制了这种反应(图5D-E)。体内实验揭示了肠道菌群改变与IL-6水平升高之间存在显著相关性(图5F-G),后续干预实验证实了DCA和FXR在促进IL-6分泌中的作用(图5H)。重要的是,使用单克隆抗体阻断IL-6/IL-6R轴可显著抑制肿瘤生长并减少G-MDSC浸润(图5I-J),表明IL-6是DCA-FXR信号通路的关键下游效应分子,该通路驱动免疫抑制性肿瘤微环境的形成。
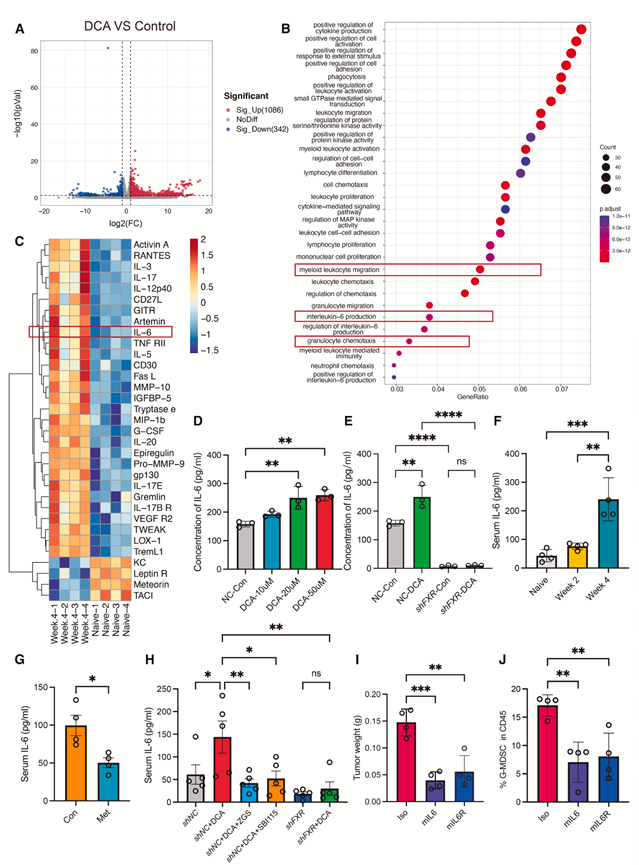
图5. IL-6 水平升高与 DCA 诱导的肿瘤免疫抑制微环境有关。
(A) 火山图显示DCA处理与对照荷瘤小鼠肿瘤组织中的差异基因。(B) 与对照组相比,DCA处理小鼠差异表达基因的GSEA。(C) 细胞因子阵列法检测幼年和4T1荷瘤小鼠血清中细胞因子的表达。(D) 在使用或不使用二氯乙酸培养 48 小时的 shNC-4T1(正常细胞)细胞所培养的培养基中,IL-6 的浓度情况。(E) 细胞培养基中IL-6的浓度。(F) 4T1荷瘤小鼠不同时期血清中IL-6水平的变化。(G) 甲硝唑对4T1荷瘤小鼠血清中IL-6水平的影响。(H) 各治疗组血清IL-6水平。(I) 用 IgG 无免疫原性对照组、中和性αIL-6 单克隆抗体(mIL-6)或中和性αIL-6R 单克隆抗体(mIL-6R)处理的 4T1 肿瘤荷瘤小鼠的肿瘤重量。(J) 不同治疗肿瘤中CD45肿瘤浸润免疫细胞中G-MDSC的流式细胞分析。
06
激活的NF-κB通路负责免疫抑制细胞的募集和分化
随后,对DCA处理组和对照组肿瘤的转录谱进行基因集富集分析(GSEA),结果显示NF-κB信号通路显著激活(图6A),该通路是IL-6表达的关键驱动因素。与此一致的是,对4T1-shNC和4T1-shFXR细胞的比较转录组分析表明,在FXR缺陷细胞中,多种NF-κB抑制基因(包括Nfkbid、Traf3、Nfkb1和Nfkbia)表达上调,而NF-κB激活基因(如Ikbkb、Traf2、Ikbkg和Traf6)在FXR完整细胞中富集。
在蛋白质水平上,与大肠杆菌相比,Lach处理以及与对照培养基RCM(强化梭菌培养基)相比,Lach培养基均导致FXR激活,并伴有p65 (p-p65)和IκB (p-IκB)磷酸化水平升高(图6B)。直接添加DCA同样以FXR依赖的方式增强NF-κB信号通路(图6C)。相反,使用FXR抑制剂ZGS或NF-κB抑制剂BAY 11-7082处理显著降低了p65和IκB的磷酸化水平,FXR敲低也产生了类似的抑制作用(图6D)。荧光素酶报告基因检测进一步证实,DCA强烈诱导NF-κB转录活性,而FXR的药理学抑制则消除了这种活性(图6E)。
为了评估 NF-κB 激活的功能后果,他们利用骨髓来源的 G-MDSC进行了体外趋化性实验,并利用脾脏 CD3+ T 细胞与经不同条件预处理的肿瘤细胞共培养进行了 Th17 极化实验。DCA 显著增强了 G-MDSC 的趋化性并促进了 Th17 分化。重要的是,FXR 或 NF-κB 的抑制,以及使用 IL-6 单克隆抗体阻断 IL-6 信号通路,均能有效消除 DCA 诱导的趋化性增强作用(图6F-I)。
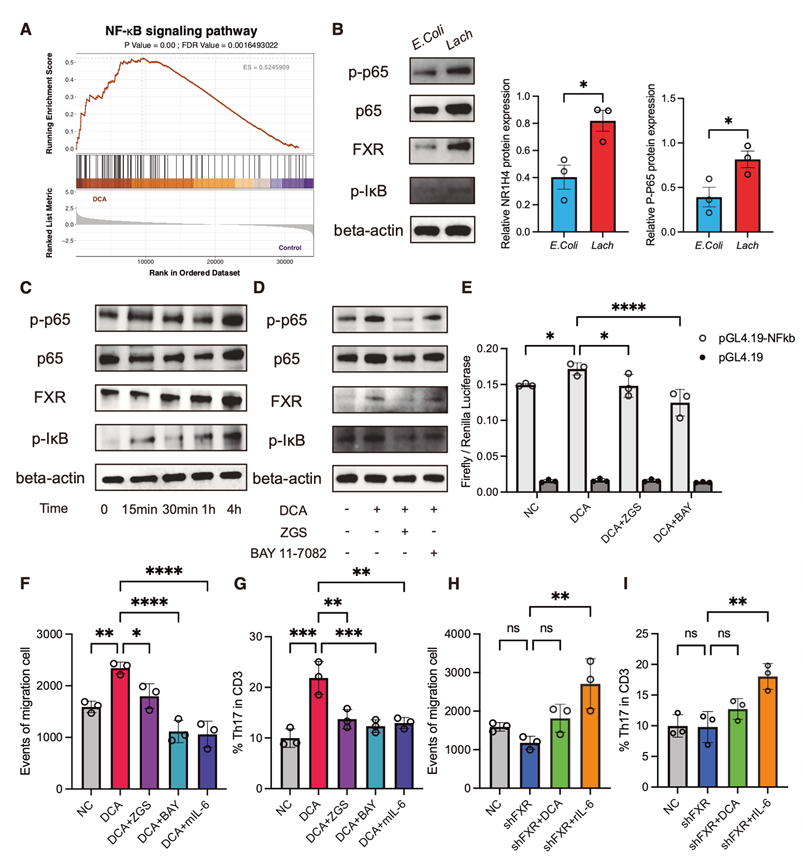
图6. 活化的NF-κB通路与免疫抑制细胞的募集和分化有关。
(A) DCA处理与对照的转录谱基因集富集分析(GSEA)。(B) Western blots的代表性图像(左)和统计分析(右)。(C) Western blot检测20 μM DCA处理4T1细胞不同时间p-p65、总p65、p- IκB和FXR的表达。(D) Western blot检测p-p65、总p65、p-IκB和FXR的表达。(E) 荧光素酶报告试验研究DCA对NF-κB活化的影响。(F) Transwell系统底部室中迁移的G-MDSC的绝对数量。(G) 共培养系统中Th17细胞的极化。(H) Transwell系统底部室中迁移的G-MDSC的绝对数量。(I) 共培养系统中Th17细胞的极化。
07
IL-6分泌水平升高预示着乳腺癌预后不良
IL-6细胞因子家族,尤其是IL-6本身,与乳腺癌进展的多个标志性特征密切相关,包括肿瘤生长、侵袭、转移、血管生成和免疫调节。在队列研究中,与TAN相比,肿瘤组织中IL-6的表达显著升高(图7A)。此外,非管腔型乳腺癌亚型(通常恶性程度更高)中IL-6的水平显著高于管腔型乳腺癌亚型(图7B)。
与之前的体外和体内观察结果一致,IL-6表达与乳腺癌组织中FXR水平呈正相关(图7C)。纳入患者预后后,进一步发现高IL-6表达与较短的无远处转移生存期(DMFS)相关(图7D)。此外,IL-6表达与MPO(一种典型的G-MDSC标志物)呈显著正相关(图7E)。对公共乳腺癌数据集的分析也表明,G-MDSC和Th17细胞特征的增强与不良临床结局相关(图7F)。这些研究结果共同表明,FXR 和 IL-6 不仅有助于形成免疫抑制性微环境,而且可能作为乳腺癌的有价值的预后生物标志物。
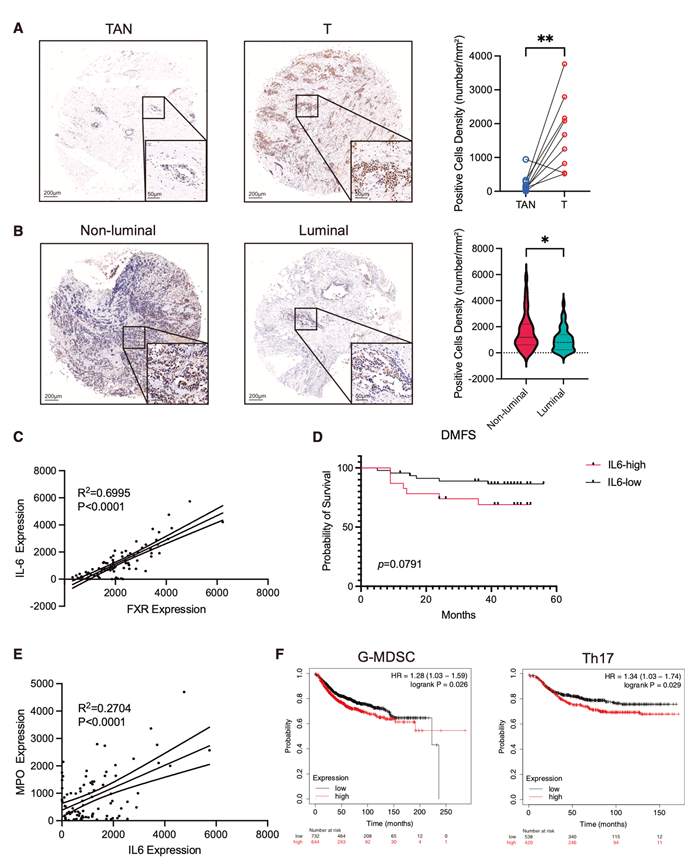
图7. IL-6分泌升高预示着乳腺癌预后不良。
(A) 代表性的图像(左)和统计分析(右)展示了患有乳腺癌的个体中配对的肿瘤组织(T)与肿瘤旁正常组织(TAN)中 IL-6 免疫组化染色的结果。(B) 乳腺癌亚型肿瘤组织中 IL-6 免疫组化染色的统计分析。(C) 乳腺癌组织中IL-6与FXR (NR1H4) IHC表达水平的Pearson相关性分析。(D) 乳腺癌患者DMFS与IL-6的Kaplan-Meier图。(E) 乳腺癌肿瘤组织中MPO与IL-6 IHC表达水平的Pearson相关性分析。(F) 利用公共转录组数据对患有乳腺癌的个体按照推断出的 G-MDSC(左图)和 Th17(右图)特征进行分层的 DMFS(疾病无进展生存率)进行 Kaplan-Meier 分析。
+ + + + + + + + + + +
结 论
本研究通过多组学分析发现,E. bolteae在肿瘤发展过程中逐渐富集,并伴随菌群来源代谢产物DCA水平的升高。DCA在肿瘤中积累,激活肿瘤细胞中的FXR,进而通过核因子κB(NF-κB)信号通路诱导IL-6的产生。IL-6促进GM-DRS和Th17细胞的募集,从而建立免疫抑制性微环境。抑制或敲低FXR以及阻断IL-6信号通路均可减弱这些作用。这些发现揭示了驱动乳腺癌进展的菌群-代谢产物-免疫轴,并提示微生物代谢产物可作为潜在的治疗靶点。
+ + + + +



